doi: 10.15389/agrobiology.2022.6.1136rus
УДК 636.5:636.064.6:577.21
Работа выполнена при финансовой поддержке Российского научного фонда, грант № 21-16-00086.
ПОЛНОГЕНОМНЫЕ АССОЦИАТИВНЫЕ ИССЛЕДОВАНИЯ ПОКАЗАТЕЛЕЙ РОСТА У ПЕРЕПЕЛОВ Coturnix japonica
Н.Ю. ГЕРМАН1, Н.А. ВОЛКОВА1 ✉, П.В. ЛАРИОНОВА1, А.Н. ВЕТОХ1, Л.А. ВОЛКОВА1, А.А. СЕРМЯГИН1, А.В. ШАХИН1, Д.В. АНШАКОВ2, В.И. ФИСИНИН3, Н.А. ЗИНОВЬЕВА1
Идентификация и картирование генов, детерминирующих проявление селекционно значимых признаков у сельскохозяйственных животных, в том числе птицы, — одна из ключевых задач геномной селекции, направленной на повышение эффективности животноводства. За последние годы с использованием полногеномного анализа ассоциаций выявлено достаточно большое число важных генов-кандидатов у разных видов сельскохозяйственных животных. При этом из многочисленных видов сельскохозяйственной птицы существенная доля исследований по поиску и идентификации локусов количественных признаков (QTL, quantitative trait loci) проведена на курах. На перепелах число подобных исследований относительно невелико. Это связано прежде всего с отсутствием коммерческих чипов, что затрудняет поиск SNP и идентификацию генов, связанных с селекционно значимыми признаками. К настоящему времени в литературе недостаточно информации о локусах количественных признаков перепелов, достоверно связанных с продуктивностью. Живая масса и скорость роста перепелов — показатели мясной продуктивности, которые зависят от условий кормления и содержания птицы и детерминированы множеством QTL. В настоящем сообщении представлены результаты полногеномных ассоциативных исследований скорости роста перепелов из F2 модельной ресурсной популяции. Целью работы была идентификация локусов количественных признаков в геноме перепелов и анализ ассоциации найденных мутаций с показателем массы тела, а также характеристика аллельных вариантов в изученной F2 модельной ресурсной популяции. Перепела F2 модельной ресурсной популяции были получены посредством серии межпородных скрещиваний японского перепела (медленный рост) и техасского перепела (быстрый рост). Генотипирование полученных особей F2 (n = 232) проводили методом GBS (genotyping by sequencing). После фильтрации при обработке данных генотипирования для последующего анализа отобрали 92686 SNP. C использованием программного обеспечения PLINK 1.9 (https://www.cog-genomics.org/plink/) с принятыми ограничениями (geno 0,1, mind 0,2, maf 0,05) изучили ассоциации между данными полногеномного генотипирования и живой массой, отражающей физическое развитие птицы. Показано, что полученная F2 ресурсная популяция перепелов характеризовалась высокой изменчивостью этого показателя. У 1-суточных перепелят живая масса варьировала от 5 до 11 г и составила в среднем 9±0,1 г. В возрасте 2, 4, 6 и 8 нед она достигала соответственно 69±1, 157±2, 219±2 и 252±2 г. На основании проведенного GWAS-анализа идентифицированы 149 SNP на 1-й, 2-й, 3-й, 5-й, 6-й, 8-й, 11-й, 14-й, 15-й, 20-й, 24-й, 25-й и 26-й хромосомах, которые с высокой достоверностью (p < 0,00001) ассоциированы с живой массой. При этом на 1-й, 2-й, 3-й,5-й, 11-й и 26-й хромосомах детектированы блоки из 2-9 SNP, относящиеся к одному гену. Установлены семь генов-кандидатов (PCDH9, SMAD9, PAN4, EGFR,WDPCP, MDGA2 и PEPD), достоверно (p < 0,00001) связанных с показателем живой массы в возрасте 8 нед. Выявленные SNP могут быть в дальнейшем изучены в качестве генетических маркеров в программах селекции на увеличение массы перепелов, а также в связи с остальными показателями продуктивности.
Ключевые слова: Coturnix japonica, перепел, QTL, SNP, GBS, GWAS, живая масса, динамика роста.
Мясо и яйцо птицы, которые на сегодняшний день составляют значительную долю в общей продукции животноводства в мире (1, 2), в основном производятся с использованием видов, относящихся к семейству Phasianidae. Особым спросом пользуется продукция перепеловодства, что объясняется высокой пищевой ценностью и вкусовыми качествами яиц и мяса перепелов, а также их скороспелостью (3-5). Потребность рынка в этих продуктах способствовала созданию крупных перепеловодческих хозяйств и интенсивному развитию отрасли, в результате чего перепелиные яйца и мясо стали продуктами повседневного спроса. Рентабельность и конкурентоспособность перепеловодства обусловлены несколькими причинами. Один из определяющих факторов — использование пород и линий птицы, характеризующихся важными селекционно значимыми признаками, из которых можно выделить высокую яичную и мясную продуктивность, устойчивость к производственным стрессам, инфекционным заболеваниям, скороспелость. Создание высокопродуктивных пород, линий и кроссов невозможно без эффективной селекционной работы по поиску и идентификации ценных генотипов с использованием современных методов и подходов, основанных на изучении молекулярно-генетических механизмов формирования и проявления селекционно значимых признаков.
У сельскохозяйственных животных для значительного числа хозяйственно полезных признаков в настоящее время идентифицированы тысячи локусов количественных признаков (QTL, quantitative trait loci) (6). Из многочисленных видов домашней птицы существенная доля исследований по поиску и идентификации QTL проведена на курах (7). В базе данных QTL кур (Chicken QTLdb, https://www.animalgenome.org/cgi-bin/QTLdb/GG/in-dex) содержится информация о 16656 QTL 370 различных признаков. Большинство таких QTL выявлены с использованием в качестве генетических маркеров как микросателлитов (8, 9), так и SNP, идентифицированных посредством генотипирования единичных точечных мутаций (10) или проведения полногеномного анализа (11, 12).
В отличие от кур, исследования на перепелах по идентификации QTL, связанных с важными селекционно значимыми признаками, сравнительно малочисленны. На сегодняшний день число таких публикаций относительно невелико. В ряде работ сообщается об идентификации QTL с использованием микросателлитов, изучена связь этих генетических маркеров с показателями роста (13-15), развития (16), качества мяса (17), показателями яичной продуктивности (18). Однако следует отметить, что при выявлении QTL с использованием микросателлитов возникают определенные трудности с идентификацией соответствующих генов. Для решения проблемы осуществляется поиск SNP, ассоциированных с селекционно значимыми признаками, но проведение таких исследований на перепелах ограничено отсутствием соответствующих коммерческих чипов для выявления SNP. В последние годы растет число работ по полногеномному генотипированию (19). С использованием такого подхода выявлены SNP и идентифицированы гены, связанные с показателями продуктивности (20), поведением (21, 22), окраской оперения (23, 24), качеством яиц (25) у перепелов.
В настоящем сообщении мы представляем результаты полногеномных ассоциативных исследований показателей роста у перепелов F2 модельной ресурсной популяции в сопоставлении с динамикой изменения живой массы птицы в возрасте от 1 сут до 8 нед. Новизна использованного нами подхода заключается в создании модельной ресурсной популяции перепелов, которая характеризуется высокой степенью вариабельности изучаемых показателей роста за счет использования разводимых на территории России материнской и отцовской пород, контрастных по изучаемым показателям (быстрорастущая и медленнорастущая породы). Выявлены новые SNP и идентифицированы гены, с высокой достоверностью (p < 0,00001) ассоциированные с показателями роста. Детектированные SNP могут быть в дальнейшем изучены в качестве генетических маркеров в программах селекции на увеличение массы перепелов, а также улучшение других показателей мясной продуктивности.
Цель работы — идентификация локусов количественных признаков в геноме перепелов и анализ ассоциации найденных мутаций с живой массой, а также характеристика аллельных вариантов в F2 полученной модельной ресурсной популяции перепелов.
Методика. Исследования проводили на базе физиологического двора ФГБНУ ФИЦ ВИЖ им. Л.К. Эрнста в 2021-2022 годах на 116 самках и 116 самцах перепелов F2 модельной ресурсной популяции, которые были получены посредством межпородного скрещивания японского и техасского перепелов. Первоначально получали межпородные помеси F1 с кровностью 50 % японского перепела и 50 % техасского перепела, далее помеси F1 скрещивали между собой. Потомство F2 использовали в качестве модельной ресурсной популяции для молекулярно-генетических исследований и оценки динамики роста.
Родительские перепела (исходные породы, F1) для получения потомства содержались группами по 5-6 гол. (1 самец, 4-5 самок) в разделенных многоярусных перепелиных клетках при плотности посадки не менее 250 см2/гол. Перепела F2 модельной ресурсной популяции содержались в многоярусных перепелиных клетках (не более 25 гол. на ярус, плотность посадки не менее 162 см2/гол.). Все клетки были оборудованы системой ниппельного поения (из расчета 1 ниппель на 10 гол.), навесными кормушками (фронт кормления составлял не менее 3 см/гол.) и системой пометоудаления, в связи с чем птица имела свободный доступ к чистой воде и полнорационному промышленныму комбикорму. Для молодняка перепелов в возрасте от 1 сут до 6 нед использовался промышленный комбикорм с пищевой ценностью 11,92 (2850) МДж/кг (Ккал/кг) и содержанием сырого протеина 285,00 г/кг. С 7-недельного возраста перепелов переводили на корм для продуктивных перепелов с обменной энергией 12,13 (2900) МДж/кг (Ккал/кг) и содержанием сырого протеина 180,00 г/кг. Температуру в помещениях, где содержалась птица, поддерживали в пределах от 18 до 25 °С. В брудерах, где содержался молодняк от вылупления до возраста 4 нед, температура менялась от 35 до 23 °С по мере взросления особей. Влажность в помещениях не превышала 70 %.
Живую массу перепелов полученной F2 модельной ресурсной популяции (n = 232) учитывали в возрастной динамике с интервалом 2 нед. Перепелов взвешивали на весах OHAUS Pioneer PA413C («OHAUS», США) в возрасте 1 сут, 2, 4, 6 и 8 нед.
ДНК из проб ткани и пульпы пера перепелов выделяли с использованием набора ДНК-Экстран 2 (ООО «НПФ Синтол», Россия) согласно протоколу, рекомендованному производителем. Концентрацию полученной ДНК определяли на флуориметре Qubit 2.0 («Invitrogen/Life Technologies», США), чистоту препарата оценивали спектрофотометрически (NanoDrop 8000, «Thermo Fisher Scientific», США) (для последующего анализа использовали ДНК с соотношением OD260/280 не ниже 1,8), качество ДНК — также посредством гель-электрофореза в 1 % агарозном геле.
Полногеномное генотипирование перепелов F2 модельной ресурсной популяции проводили методом GBS (genotype by sequencing) (26). Полученная в результате GBS анализа матрица молекулярных данных, включающая секвенированные последовательности геномной ДНК 232 особей F2, после преобразования в пригодный для анализа формат, которое включало удаление адаптеров и демультиплексирование файла fastq (разделение по образцам) с использованием программы cutadapt (version 3.4, https://pypi.org/project/cutadapt/), контроль качества файлов fastq в программе fastqc (version 0.9.11, https://github.com/s-andrews/FastQC), выравнивание с использованием референсного генома с помощью пакета bowtie2 (version 2.4.4, https://github.com/BenLangmead/bowtie2), вызов вариантов (SNP) и аннотацию в пакете bcftools (version 1.13, https://samtools.gith-ub.io/bcftools/bcftools.html) была загружена в R-пакет (27) для выполнения дальнейших расчетов. В качестве референсного использовали геном Japanese quail (Coturnix japonica 2.1, GCF_001577835.2). Было отобрано 115906 SNP, из которых после оценки контроля качества и фильтрации данных генотипирования с применением программного пакета PLINK 1.9 (http://zzz.bwh.har-vard.edu/plink/) для дальнейшего GWAS (whole-genome associated study) использовали 92686 SNP. При отборе SNP учитывали качество генотипирования по всем исследуемым SNP для каждой особи (не ниже 90 %), частоту встречаемости минорных аллелей, отклонение частоты SNP генотипов от распределения Харди-Вайнберга в совокупности протестированных образцов с достоверностью p-value < 10-5.
Анализ главных компонент (PCA, https://www.datacamp.com/tuto-rial/pca-analysis-r) проводили и визуализировали в пакете R ggplot2 (https://gith-ub.com/tidyverse/ggplot2). Файлы данных были подготовлены в программной среде R4.0.
Для выявления ассоциаций SNP с показателем живой массы перепелов применяли регрессионный анализ в PLINK 1.9. Достоверность влияния SNP и определение значимых регионов в геноме перепелов оценивали с использованием теста для проверки нулевых гипотез по Бонферрони при пороговом значении p < 1×10-5. Данные визуализировали в пакете qqman с помощью языка программирования R (27). Поиск генов-кандидатов, расположенных в области идентифицированных SNP, осуществляли с помощью геномного ресурса Genome assembly Coturnix japonica 2.1 (https://www.ncbi.nlm.nih.gov/data-hub/genome/GCF_001577835.2/, дата обращения 10.07.2022).
Статистические показатели рассчитывали многофакторным дисперсионным анализом в программе Microsoft Excel 2013 по следующим параметрам: среднее арифметическое (М) и стандартная ошибка (±SEM), минимум (min), максимум (max), медиана (Мe), критерий согласия Пирсона (χ2), коэффициент гомозиготности по Робертсону (Ca), уровень полиморфизма по Робертсону при 2 аллелях (Na).
Результаты. Создание F2 ресурсной популяции с использованием контрастных по генотипу и фенотипу пород в качестве исходных родительских форм позволяет на относительно небольшом поголовье птицы получать популяции особей, характеризующиеся существенно большим размахом изменчивости по ряду фенотипических признаков. У чистопородных особей одной породы или линии отмечается, как правило, снижение изменчивости в результате направленной селекции по ограниченному числу признаков, что в случае использования такой птицы в молекулярно-генетических исследованиях по идентификации QTL требует сбора и анализа фенотипических данных на значительной выборке особей. Работы по созданию и использованию F2 модельных ресурсных популяций для молекулярно-генетических исследований широко проводятся на различных видах сельскохозяйственной птицы, в частности курах (28), индейках (29), перепелах (19-21). У перепелов в качестве исходных родительских форм во многих исследованиях используются контрастные по изучаемому признаку линии одной породы — японского перепела (19-21). В наших исследованиях модельная ресурсная популяция была получена при межпородном скрещивании японского и техасского перепелов. Японский перепел относится к яичной породе и характеризуется относительно невысокой скоростью роста, техасский перепел — мясная порода с высокой скоростью прироста живой массы.
Живая масса 1-суточных перепелов F2 модельной ресурсной варьировала от 5 до 11 г и составила в среднем 9±0,1 г. В возрасте 2, 4, 6 и 8 нед этот показатель увеличивался соответственно в 8, 17, 24 и 28 раз относительно установленного для 1-суточных перепелят (табл. 1).
Следует отметить высокую вариабельность и значительный разброс по величине живой массы у птицы в популяции F2. На рисунке 1 представлена диаграмма, отражающая распределение перепелов исследуемой выборки по этому показателю в разные возрастные периоды. Видно, что интервал вариабельности составлял 25-40 % по максимальному значению и 27-54 % — по минимальному. При этом мы установили отсутствие значительного смещения медианы по показателю живой массы у перепелов F2 по возрастным периодам наблюдения, что свидетельствует об относительно равномерном распределении частот встречаемости максимальных и минимальных значений признака в изучаемой популяции.
Неоднородность F2 модельной ресурсной популяции перепелов по живой массе подтвердил анализ главных компонент (PCA, Principal Component Analysis) на основе данных о нуклеотидных последовательностях, полученных при индивидуальном полногеномном анализе особей с использованием метода GBS (рис. 2). Первая компонента отвечает за 13,39 %, вторая — за 6,43 % генетических различий птицы в исследуемой выборке. Согласно рисунку 2, изученная популяция была представлена пятью кластерами, образующими три ветви, расходящиеся в пространстве из единой точки. Гетерогенность выборки может быть обусловлена структурой популяции, состоящей из семейств, где имеется большее генетическое сходство между особями, относящимися к одной группе, чем в среднем по популяции.
Для проверки предположения о геномной обусловленности показателей продуктивности у перепелов F2 (в частности, живой массы) мы провели полногеномный анализ ассоциаций (GWAS, whole-genome associated study). GWAS-анализ дает возможность идентифицировать множество ге-номных вариантов, связанных с продуктивными признаками птицы. В результате проведенного анализа мы выявили 149 SNP с высокой достоверностью (p < 0,00001) на 1-й, 2-й, 3-й, 5-й, 6-й, 8-й, 11-й, 14-й, 15-й, 20-й, 24-й, 25-й, 26-й хромосомах, при этом на 1-й, 2-й, 3-й, 5-й, 11-й, 26-й хромосомах были детектированы блоки из 2-9 SNP, относящиеся к одному гену. Соотношение найденных в исследовании мутаций с SNP, которые высокодостоверно ассоциированы с живой массой, и SNP, которые охарактеризованы по механизму влияния, графически представлено на рисунке 3.
Локусы SNP (p < 0,00001), выявленные в геноме перепелов в возрасте 8 нед и ассоциированные с показателем живой массы, мы использовали для идентификации позиционных и функциональных генов-кандидатов, связанных с каким-либо механизмом влияния на процессы в организме (табл. 2). В результате в базе данных NCBI (Coturnix japonica 2.1) вблизи значимых SNP обнаружили семь функциональных генов-кандидатов на 1-й (PCDH9, SMAD9, PAN4), 2-й (EGFR), 3-й (WDPCP), 5-й (MDGA2) и 11-й (PEPD) хромосомах.
Согласно данным литературы, у перепелов ген EGFR отвечает за регуляцию пролиферации клеток гранулезы и влияет на развитие фолликулов (24). Остальные мутации упоминались в литературе в отношении других животных и птицы как ортологические группы генов (30), определенные биоинформатически с использованием комбинации сходства последовательностей белков и данных о локальной синтении.
Были рассчитаны частоты встречаемости, степень полиморфизма и генетическое равновесие по выявленным генам в изученной популяции перепелов F2 в возрасте 8 нед (табл. 3).
Согласно полученным результатам, по всем детектированным точечным мутациям в генах имелись полиморфные варианты генотипов с вариацией степени гомозиготности (по Робертсону) от 0,509 до 0,893. Выявлены отклонения от генетического равновесия для генов PAN4 (7,650), EGFR_1 (5,603), EGFR_2 (4,115) и WDPCP (20,819).
Анализ открытых информационных источников показал, что на сегодняшний день опубликовано небольшое число работ, касающихся поиска и идентификации QTL, которые связаны с показателями роста у перепелов. Так, M.I. Haqani с соавт. (19) определили QTL, ассоциированные с показа-телем живой массы у перепелов, исследовав возрастную динамику этого показателя у 277 особей F2, полученных при скрещивании линий японского перепела с повышенной и нормальной живой массой. Живую массу определяли еженедельно от вылупления до возраста 16 нед. В результате установили 125 SNP, ассоциированных с живой массой. На 1-й и 3-й хромосомах были выявлены 4 QTL, связанные с живой массой птицы в возрасте от 4 до 16 нед. При этом в ранний возрастной период (до 3 нед) статистически значимых QTL не обнаружили (19). S. Vollmar соавт. (20) на 920 японских перепелах F2 провели идентификацию QTL, ассоциированных с использованием фосфора, кальция, конверсией корма и приростом живой массы, который рассчитывали на основании данных о живой массе в возрасте 10 и 15 сут. Для анализа отобрали 3986 SNP. В результате авторы установили 12 значимых SNP и идентифицировали 4 гена-кандидата, которые были связаны с изучаемыми признаками (20). J. Recoquillay с соавт. (21) получили F2 модельную ресурсную популяцию японского перепела скрещиванием двух линий, контрастных по особенностям поведения. Наряду с поведенческими реакциями у птицы из модельной популяции изучили продуктивные признаки, в частности определили живую массу в возрасте 17 и 65 сут и яйценоскость. На основании полногеномного генотипирования обнаружили 22 QTL, связанных с продуктивными признаками. Выявленные SNP были идентифицированы на 1-й, 3-й, 5-й, 8-й, 10-й и 18-й хромосомах (21).
В наших исследованиях одной из родительских форм при создании ресурсной популяции также был японский перепел. При этом использование в качестве контрастной породы техасского перепела позволило получить F2 популяцию со значительным размахом изменчивости по живой массе. Новизна проведенного нами исследования заключается в идентификации не описанных ранее в открытых информационных источниках SNP и генов-кандидатов, ассоциированных с показателем живой массы у перепелов в разные возрастные периоды. Расположенные вблизи функциональных генов достоверно (p < 0,00001) значимые SNP, связанные с показателем живой массы перепелов в возрасте до 8 нед, были локализованы на 13 из 28 учтенных хромосом, в том числе на 1-й, 3-й, 5-й и 8-й хромосомах, что аналогично данным, полученным другими исследователями (19, 21). Наибольшее число таких SNP локализовались на 1-й, 2-й, 3-й, 15-й и 27-й хромосомах (см. рис. 3).
Таким образом, выполненные нами исследования полногеномных последовательностей ДНК в F2 полученной модельной ресурсной популяции перепелов (японский перепел × техасский перепел) позволили обнаружить на 1-й, 2-й, 3-й, 5-й, 6-й, 8-й, 11-й, 14-й, 15-й, 20-й, 24-й, 25-й и 26-й хромосомах 149 SNP, ассоциированных (p < 0,00001) с живой массой. При этом на 1-й, 2-й, 3-й, 5-й, 11-й и 26-й хромосомах были детектированы блоки из 2-9 SNP, относящиеся к одному гену. В районах нахождения выявленных локусов количественных признаков расположены несколько позиционных и функциональных маркерных генов-кандидатов, задействованных в проявлении признаков, отличных от изучаемого, — живой массы. В частности, установлены семь генов-кандидатов (PCDH9, SMAD9, PAN4, EGFR, WDPCP, MDGA2 и PEPD), достоверно (p < 0,00001) связанных с показателем живой массы в возрасте 8 нед. Полученные данные составят основу для продолжения исследований по поиску ассоциаций выявленных мутаций с другими селекционно значимыми показателями продуктивности.
1. Динамика роста перепелов F2 модельной ресурсной популяции (японский перепел × техасский перепел) |
|||||||
Возраст |
Живая масса, г |
||||||
абсолютная |
прирост |
||||||
средняя |
ошибка |
максимум |
минимум |
медиана |
абсолютный |
среднесуточный |
|
1 сут |
9 |
0,1 |
11 |
5 |
9 |
|
|
2 нед |
69 |
0,9 |
117 |
32 |
69 |
61 |
9 |
4 нед |
157 |
1,5 |
214 |
103 |
159 |
88 |
13 |
6 нед |
219 |
1,9 |
322 |
159 |
217 |
62 |
9 |
8 нед |
252 |
2,2 |
354 |
163 |
248 |
33 |
5 |
2. SNP и гены-кандидаты, ассоциированные с показателями живой массы у перепелов из F2 модельной ресурсной |
||||
Хромосома |
Число SNP |
SNP, позиция bp |
Достоверность p |
Ген |
1-я |
3 |
142741390 |
7,25×10-8 |
PCDH9 |
|
|
154153381 |
1,04×10-5 |
SMAD9 |
|
|
157425751 |
1,35×10-5 |
PAN4 |
2-я |
2 |
73896881 |
1,23×10-9 |
EGFR |
|
|
73896952 |
3,67×10-8 |
|
3-я |
1 |
1993165 |
6,77×10-10 |
WDPCP |
5-я |
2 |
51581638 |
6,71×10-5 |
MDGA2 |
|
|
51581710 |
6,71×10-5 |
|
11-я |
1 |
9015455 |
8,51×10-5 |
PEPD |
3. Оценка частоты встречаемости селекционно значимых генетических маркеров у перепелов из F2 модельной ресурсной популяции (японский перепел × техасский перепел) (n = 232, физиологический двор, ВИЖ им. академика Л.К. Эрнста, 2021-2022 годы) |
|||||||||
Хромо-сома |
Ген |
Частота генотипов, M±SEM |
Частота аллелей |
χ2 |
Ca |
Na |
|||
11, Н и О |
12, Н и О |
22, Н и О |
1 |
2 |
|||||
1-я |
SMAD9 |
0,178±0,019 |
0,511±0,024 |
0,311±0,022 |
0,434 |
0,566 |
0,120 |
0,509 |
1,966 |
|
0,188 |
0,491 |
0,321 |
||||||
|
PSDH9 |
0,018±0,006 |
0,283±0,021 |
0,699±0,022 |
0,160 |
0,840 |
0,455 |
0,731 |
1,367 |
|
0,026 |
0,269 |
0,706 |
||||||
|
PAN4 |
0,023±0,007 |
0,502±0,024 |
0,474±0,024 |
0,275 |
0,725 |
7,650 |
0,602 |
1,662 |
|
0,075 |
0,398 |
0,526 |
||||||
2-я |
EGFR_1 |
0 |
0,320±0,021 |
0,680±0,022 |
0,160 |
0,840 |
5,603 |
0,731 |
1,367 |
|
0,026 |
0,269 |
0,706 |
||||||
|
EGFR_2 |
0 |
0,274±0,020 |
0,726±0,021 |
0,137 |
0,863 |
4,115 |
0,764 |
1,310 |
|
0,019 |
0,236 |
0,745 |
||||||
3-я |
WDPCP |
0,065±0,003 |
0,159±0,021 |
0,776±0,021 |
0,145 |
0,855 |
20,189 |
0,752 |
1,329 |
|
0,021 |
0,248 |
0,731 |
||||||
5-я |
MDGA |
0 |
0,205±0,019 |
0,795±0,019 |
0,103 |
0,897 |
2,314 |
0,816 |
1,226 |
|
0,011 |
0,184 |
0,805 |
||||||
11-я |
PEPD_2 |
0,009±0,003 |
0,095±0,015 |
0,896±0,015 |
0,057 |
0,943 |
2,545 |
0,893 |
1,120 |
|
0,003 |
0,107 |
0,889 |
||||||
Примечание. Для частоты генотипов и аллелей приведены варианты кодирования (11 — аллели 1 и 1, 12 — аллели 1 и 2, 22 — аллели 2 и 2); χ2 — критерий согласия Пирсона, Ca — коэффициент гомозиготности по Робертсону, Na — уровень полиморфизма по Робертсону при 2 аллелях, Н и О — соответственно наблюдаемая и ожидаемая частота. |
|||||||||
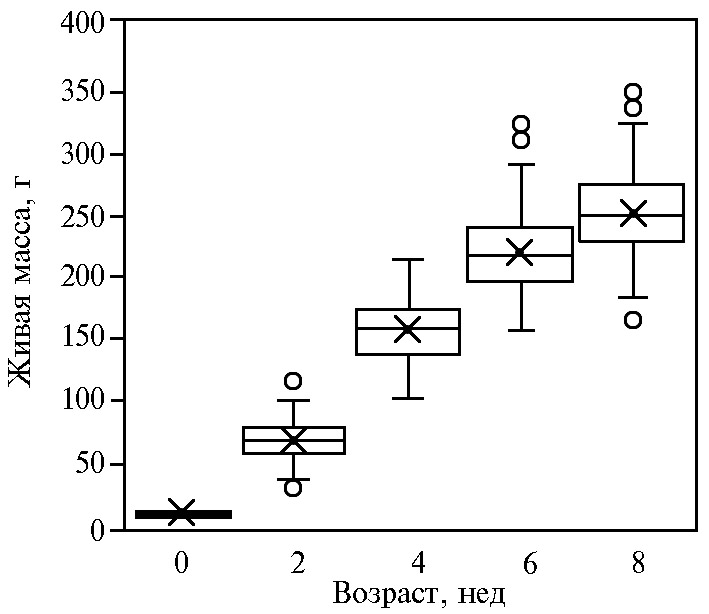
Рис. 1. Характеристика F2 модельной ресурсной популяции перепелов (японский перепел × техасский перепел) по показателю живой массы в разные возрастные периоды (n = 232, физиологический двор, ВИЖ им. академика Л.К. Эрнста, 2021-2022 годы).
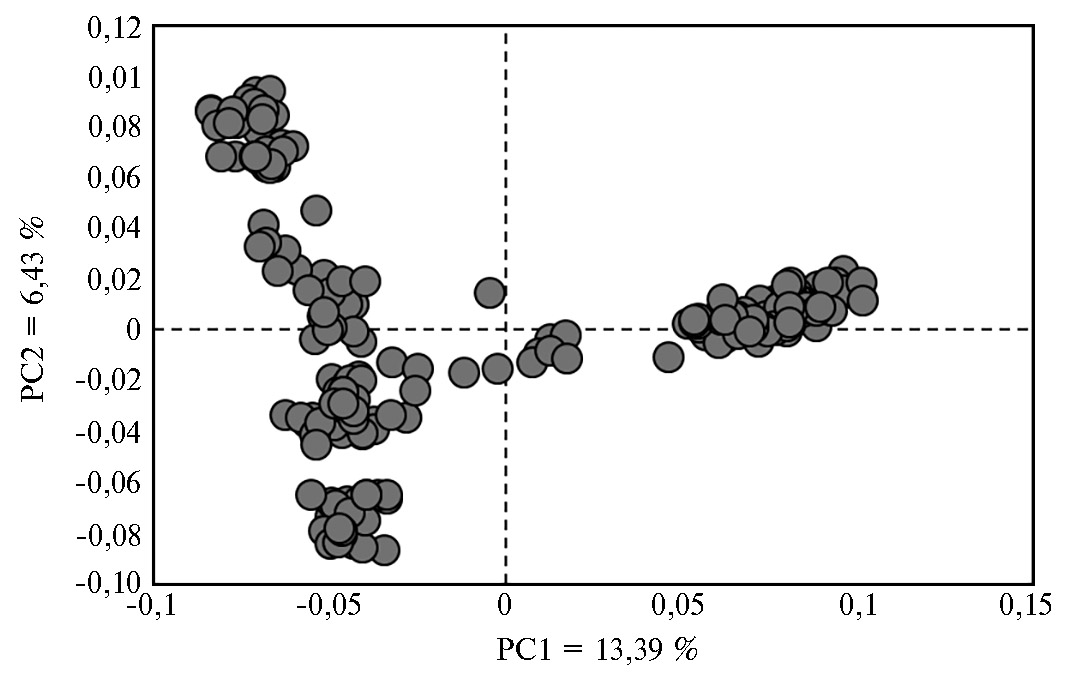
Рис. 2. PCA-aнализ данных полногеномного GBS (genotype by sequencing) генотипирования особей из F2 модельной ресурсной популяции перепелов (японский перепел × техасский перепел) (n = 232, физиологический двор, ВИЖ им. академика Л.К. Эрнста, 2021-2022 годы).
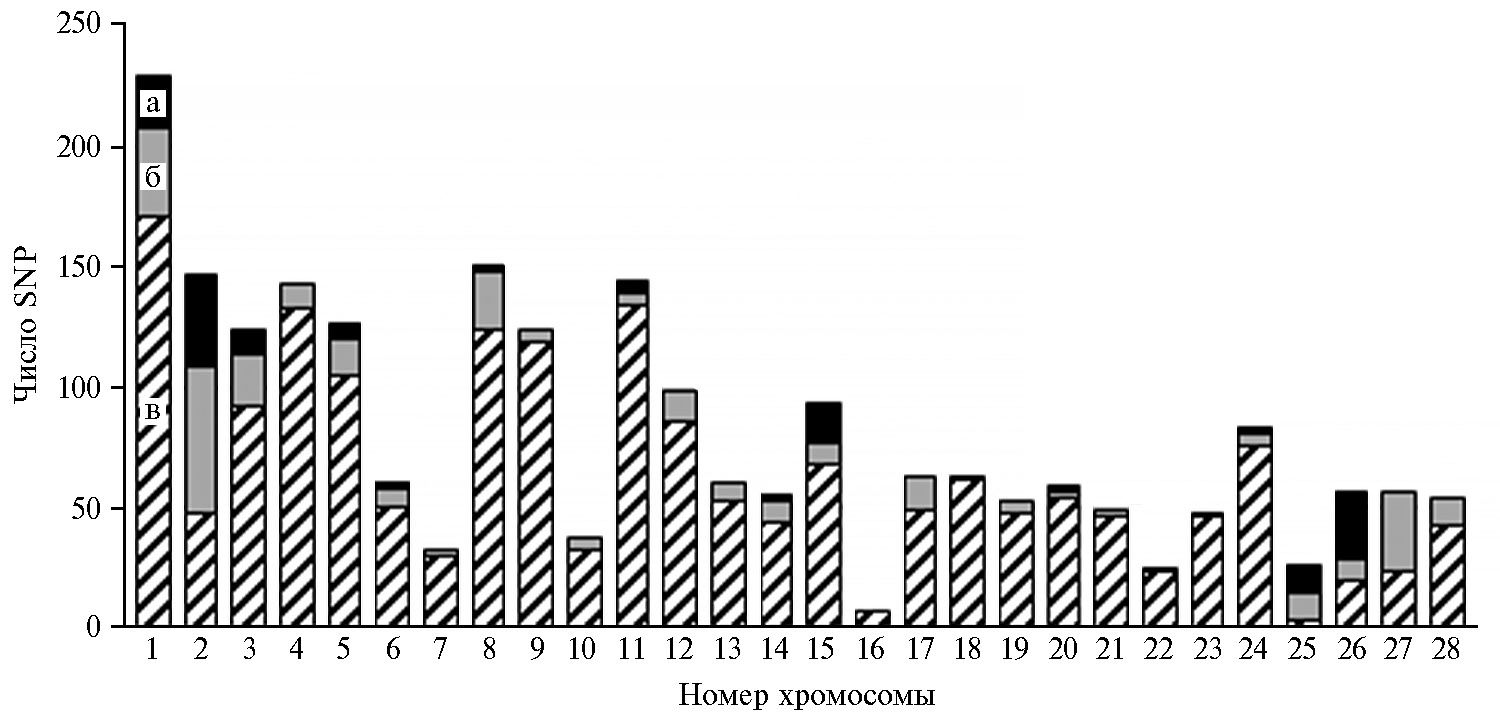
Рис. 3. Характеристика SNP, ассоциированных с показателями живой массы, у перепелов из F2 модельной ресурсной популяции (японский перепел × техасский перепел) в возрасте от 2 до 8 нед: а — охарактеризованные (расположенные вблизи функциональных генов) SNP с достоверностью ассоциаций выше 1×10-5, б — неохарактеризованные (не локализованы вблизи функциональных генов) SNP с достоверностью ассоциаций выше 1×10-5, в — SNP с достоверностью ассоциаций ниже 1×10-5 (n = 232, физиологический двор, ВИЖ им. академика Л.К. Эрнста, 2021-2022 годы).
ЛИТЕРАТУРА
- Фисинин В.И., Буяров В.С., Буяров А.В., Шуметов В.Г. Мясное птицеводство в регионах России: современное состояние и перспективы инновационного развития. Аграрная наука, 2018, 2:30-38.
- Зыков С.А. Современные тенденции развития птицеводства. Эффективное животноводство, 2019, 4(152): 51-54.
- Lukanov H. Domestic quail (Coturnix japonica domestica), is there such farm animal? World's Poultry Science Journal, 2019, 75(4): 547-558 (doi: 10.1017/S0043933919000631).
- Lukanov H., Pavlova I. Domestication changes in Japanese quail (Coturnix japonica): a review. World'sPoultryScienceJournal, 2020, 76(4): 787-801 (doi: 10.1080/00439339.2020.1823303).
- Крапчина Л.Н., Гемаюрова К.С. Перепеловодство как перспективный вид предпринимательской деятельности. Российское предпринимательство, 2013, 5(227): 84-89.
- Hu Z.-L., Park C.A., Reecy J.M. Bringing the Animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Research, 2022, 50(D1): D956-D961 (doi: 10.1093/nar/gkab1116).
- Goto T., Tsudzuki M. Genetic mapping of quantitative trait loci for egg production and egg quality traits in chickens: a review. J. Poult. Sci., 2017, 54(1): 1-12 (doi: 10.2141/jpsa.0160121).
- Schreiweis M.A., Hester P.Y., Settar P., Moody D. E. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Animal Genetic, 2006, 37(2): 106-112 (doi: 10.1111/j.1365-2052.2005.01394.x).
- Campos R.L.R., Ambo M., Rosario M.F., Moura A.S.A.M.T., Boschiero C., Nones K., Ledur M.C., Coutinho L.L. Potential association between microsatellite markers on chicken chromosomes 6, 7 and 8 and body weight. International Journal of Poultry Science, 2009, 8(7): 696-699 (doi: 10.3923/ijps.2009.696.699).
- Баркова О.Ю. Ассоциация однонуклеотидной замены SNP2-1 с признаками качества яйца у кур-несушек. Птицеводство, 2019, 7-8: 14-18 (doi: 10.33845/0033-3239-2019-68-7-8-14-18).
- Moreira G.C.M., Poleti M.D., Pértille F., Boschiero C., Cesar A.S.M., Godoy T.F., Ledur M.C., Reecy J.M., Garrick D.J., Coutinho L.L. Unraveling genomic associations with feed efficiency and body weight traits in chickens through an integrative approach. BMC Genet., 2019, 20(1): 83 (doi: 10.1186/s12863-019-0783-3).
- Li W., Zheng M., Zhao G., Wang J., Liu J., Wang S., Feng F., D. Liu, D. Zhu, Q. Li, Guo L., Guo Yu., Liu R., Wen J. Identification of QTL regions and candidate genes for growth and feed efficiency in broilers. Genetics Selection Evolution, 2021, 53(1): 13 (doi: 10.1186/s12711-021-00608-3).
- Ori R.J., Esmailizadeh A.K., Charati H., Mohammadabadi M.R., Sohrabi S.S. Identification of QTL for live weight and growth rate using DNA markers on chromosome 3 in an F2 population of Japanese quail. Molecular Biology Reports, 2014, 41: 10491057 (doi: 10.1007/s11033-013-2950-3).
- Sohrabi S.S., Esmailizadeh A.K., Baghizadeh B., Hasan M., Mohammad M., Nahid A., Ehsan N. Quantitative trait loci underlying hatching weight and growth traits in an F2 intercross between two strains of Japanese quail. Animal Production Science, 2012, 52(11): 1012-1018 (doi: 10.1071/AN12100).
- Esmailizadeh A.K. Baghizadeh A., Ahmadizadeh M. Genetic mapping of quantitative trait loci affecting bodyweight on chromosome 1 in a commercial strain of Japanese quail. Animal Production Science, 2011, 52(1): 64-68 (doi: 10.1071/AN11220).
- Nasirifar E., Talebi M., Esmailizadeh A.K., Moradian H., Sohrabi S.S., Askari N. A chromosome-wide QTL mapping on chromosome 2 to identify loci affecting live weight and carcass traits in F2 population of Japanese quail. Czech J. Anim. Sci., 2016, 61: 290-297 (doi: 10.17221/113/2014-CJAS).
- Tavaniello S., Maiorano G., Siwek M., Knaga S., Witkowski A., Di Memmo D., Bednarczyk M. Growth performance, meat quality traits, and genetic mapping of quantitative trait loci in 3 generations of Japanese quail populations (Coturnix japonica). Poultry Science, 2014, 93(8): 2129-2140 (doi: 10.3382/ps.2014-03920).
- Knaga S., Siwek M., Tavaniello S., Maiorano G., Witkowski A., Jeżewska-Witkowska G., Bednarczyk M., Zięba G. Identification of quantitative trait loci affecting production and biochemical traits in a unique Japanese quail resource population. Poultry Science, 2018, 97(7): 2267-2277 (doi: 10.3382/ps/pey110).
- Haqani M.I., Nomura S., Nakano M., Goto T., Nagano A.J., Takenouchi A., Nakamura Y., Ishikawa A., Tsudzuki M. Quantitative trait loci for growth-related traits in Japanese quail (Coturnix japonica) using restriction-site associated DNA sequencing. Molecular Genetics and Genomics, 2021, 296(5): 1147-1159 (doi: 10.1007/s00438-021-01806-w).
- Vollmar S., Haas V., Schmid M., Preuß S., Joshi R., Rodehutscord M., Bennewitz J. Mapping genes for phosphorus utilization and correlated traits using a 4k SNP linkage map in Japanese quail (Coturnix japonica). Animal Genetis, 2020, 52: 90-98 (doi: 10.1111/age.13018).
- Recoquillay J., Pitel А., Arnould С., Leroux S., Dehais P., Moréno C., Calandreau L., Bertin A., Gourichon D., Bouchez O., Vignal A., Fariello M. I., Minvielle F., Beaumont C., Leterrier C., Le Bihan-Duval E. A medium density genetic map and QTL for behavioral and production traits in Japanese quail. BMC Genomics, 2015, 16: 10 (doi: 10.1186/s12864-014-1210-9).
- Morris K.M., Hindle M.M., Boitard S., Burt D.W., Danner A.F., Eory L., Forrest H.L., Gourichon D., Gros J., Hillier L.W., Jaffredo Th., Khoury H., Lansford R., Leterrier C., Loudon A., Mason A.S., Meddle S.L., Minvielle F., Minx P., Pitel F., Seiler J.P., Shimmura T., Tomlinson C., Vignal A., Webster R.G., Yoshimura T., Warren W.C., Smith J. The quail genome: insights into social behaviour, seasonal biology and infectious disease response. BMC Biology, 2020, 18: 14 (doi: 10.1186/s12915-020-0743-4).
- Wu Y., Zhang Y., Hou Z., Hou Z., Fan G., Pi J., Sun S., Chen J., Liu H., Du X., Shen J., Hu G., Chen W., Pan A., Yin P., Chen X., Pu Y., Zhang H., Liang Z., Jian J., Zhang H., Wu B., Sun J., Chen J., Tao H., Yang T., Xiao H., Yang H., Zheng C., Bai M., Fang X., Burt D.W., Wang W., Li Q., Xu X., Li C., Yang H., Wang J., Yang N., Liu X., Du J. Population genomic data reveal genes related to important traits of quail. GigaScience, 2018, 7(5): giy049 (doi: 10.1093/gigascience/giy049).
- Wu Y., Xiaoet H., Pi J., Zhang H., Pan A., Pu Yu., Liang Z., Shen J., Du J. EGFR promotes the proliferation of quail follicular granulosa cells through the MAPK/extracellular signal-regulated kinase (ERK) signaling pathway. Cell Cycle, 2019, 18(20): 2742-2756 (doi: 10.1080/15384101.2019.1656952).
- Haqani M., Nomura S., Nakano M., Goto T., Nagano A.J., Takenouchi A., Nakamura Y., Ishikawa A., Tsudzuki M. Mapping of quantitative trait loci controlling egg-quality and -production traits in Japanese quail (Coturnix japonica) using restriction-site associated DNA sequencing. Genes, 2021, 12(5): 735 (doi: 10.3390/genes12050735).
- Perea C., De La Hoz J.F., Cruz D., Lobaton J.D., Izquierdo P., Quintero J.C., Raatz B., Duitama J. Bioinformatic analysis of genotype by sequencing (GBS) data with NGSEP. BMC Genomics, 2016, 17(S.5): 498 (doi: 10.1186/s12864-016-2827-7).
- Turner S.D. qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. Journal of Open Source Software, 2018, 3(25): 731 (doi: 10.21105/joss.00731).
- Schreiweis M.A., Hester P.Y., Settar P., Moody D.E. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Animal Genetics, 2006, 37(2): 106-112 (doi: 10.1111/j.1365-2052.2005.01394.x).
- Aslam M.L., Bastiaansen J.W., Crooijmans R.P., Vereijken A., Groenen M. Whole genome QTL mapping for growth, meat quality and breast meat yield traits in turkey. BMC Genetics, 2011, 12: 61 (doi: 10.1186/1471-2156-12-61).
- Simao F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 2015, 31(19): 3210-3212 (doi: 10.1093/bioinformatics/btv351).