doi: 10.15389/agrobiology.2020.2.257rus
УДК 636.2:636.082:577.21
Исследования выполнены при поддержке Минобрнауки России (тема № AAAA-A18-118021590134-3) и Минсельхоза России (тема № АААА-А19-119031590044-3).
ОЦЕНКА ГЕНОМНОЙ ВАРИАБЕЛЬНОСТИ ПРОДУКТИВНЫХ ПРИЗНАКОВ У ЖИВОТНЫХ ГОЛШТИНИЗИРОВАННОЙ ЧЕРНО-ПЕСТРОЙ ПОРОДЫ НА ОСНОВЕ GWAS АНАЛИЗА И ROH ПАТТЕРНОВ
А.А. СЕРМЯГИН1, О.А. БЫКОВА2, О.Г. ЛОРЕТЦ2, О.В. КОСТЮНИНА1, Н.А. ЗИНОВЬЕВА1
Для рационального использования накапливаемой информации племенного учета в стадах молочного скота требуется создание эталонных, или референтных, групп животных, имеющих высокую достоверность оценки племенной ценности. Это необходимо при генетической оценке, а также при геномной селекции. Однако без качественного фенотипирования создание таких групп невозможно. Нужны данные о геномной вариабельности признаков фенотипа у животных разных генераций с использованием результатов полногеномного анализа (genome-wide association study, GWAS) и паттернов гомозиготности (runs of homozygosity, ROH). Высокопродуктивное стадо голштинизированного черно-пестрого скота Урала представляет собой релевантный объект как для модельных исследований, так и для пополнения референтной популяции скота России. Новизна выполненной нами работы заключается в оценке динамики изменчивости геномного инбридинга (FROH) в поколениях животных (мать матери—мать—дочь) и его сопоставлении с непосредственной геномной племенной ценностью (direct genomic values, DGV), а также в поиске новых механизмов подтверждения научной гипотезы об использовании ограниченного набора экспериментальных данных в GWAS. Объектом исследований были 76 коров и телок голштинизированной черно-пестрой породы, а также 9 голштинских быков-производителей, генотипированных примерно по 139 тыс. SNP (single nucleotide polymorphism) на платформе Bovine GGP HD («Illumina/Neogen», США). Для расчета DGV по выборке животных использовалась российская референтная группа скота (591 гол.). GWAS и ROH анализы проведены на основе 110448 SNP. Достоверность (p-values) полногеномных ассоциаций с прямыми фенотипами коров колебалась от 2,31×10-5 до 1,08×10-7. Обнаружены локусы количественных признаков на аутосомах BTA1, BTA2, BTA5, BTA7, BTA8, BTA10, BTA11, BTA12, BTA14, BTA16, BTA20, BTA21 и BTA26. Для величины удоя детектирован регион на BTA14 (1,44-1,59 Mb) с генами ZNF16, ARHGAP39 и ZNF7, сопряженными с повышенным выходом молочного жира. Для числа осеменений выявлен ряд SNP, локализованных в генах (ARHGAP31) либо в непосредственной близости от них (SERPINA5) и связанных с интенсивностью развития до половозрелого состояния, а также овариальной функцией у животных. Приведена характеристика ROH в зависимости от длины их фрагментов в геноме. Определены консервативные гомозиготные участки на хромосомах BTA12, BTA14, BTA26 и BTA29 с входящими в них наиболее значимыми генами, которые потенциально связаны с давлением отбора в исследуемой популяции преимущественно по признакам молочной продуктивности, репродукции и качественным параметрам оценки вымени. Величина FROH достоверно (р < 0,05-0,001) увеличивалась в поколениях потомок—родитель: на +0,012, или 1,2 %, для матерей и на +0,029 для дочерей. Наибольшие значения геномного инбридинга (FROH = 0,135) отмечены для быков-отцов коров (поколение матерей) и телок (поколение дочерей). Каждая последующая генерация особей показывала среднее увеличение DGV по удою на +94,2 кг, по количеству молочного жира на +4,4 кг и белка — на +3,0 кг, что достаточно четко отражает стратегию на улучшение признаков молочной продуктивности в стаде при получении новых генотипов скота. Таким образом, показано, что на основе поиска ассоциаций и локусов, находящихся под селекционным давлением, можно оценить изменчивость прямых фенотипов крупного рогатого скота, используя ее как модель для исследований геномной вариабельности в отдельно взятом стаде.
Ключевые слова: крупный рогатый скот, GWAS-анализ, ROH паттерны, референтная популяция, геномный инбридинг, геномная оценка, молочная продуктивность, фертильность.
Методы совершенствования популяций животных за последнее столетие претерпели изменения от отбора особей по предкам и фенотипу к оценке по качеству потомства, а в последние десятилетия — по совокупности генетических маркеров. Развитие подходов геномной селекции базируется на точной фенотипической информации по хозяйственно-биологическим признакам. Поэтому эффективность дальнейших мероприятий по улучшению генетического потенциала в молочном скотоводстве зависит как от качества племенного учета, так и от выбранной стратегии разведения животных в популяции. Массивы данных, накапливаемые в стадах крупного рогатого скота, ежегодно возрастают. К родословной и фенотипу животного прибавляется его геномный паспорт, который позволяет уточнить происхождение особи и ее генетическую ценность (1, 2).
Как в России, так и в мире активно изучается геномная архитектура признаков продуктивности у животных. Характеристика изменчивости количественных и качественных параметров при этом может измеряться в генетических, фенотипических и скорректированных на ряд регрессионных факторов компонентах. Так, полногеномные ассоциативные исследования (genome-wide association study, GWAS), проведенные ранее на популяции голштинизированного черно-пестрого скота в Московской и Ленинградской областях, показали возможность применения оценок племенной ценности (еstimated breeding values, EBV) для поиска однонуклеотидных полиморфизмов (single nucleotide polymorphism, SNP), ассоциированных с локусами количественных признаков (quantitative trait loci, QTL). Были обнаружены значимые мутации в генах DGAT1 (p = 6,8×10-22), PLEC (p = 6,9×10-20)и GRINA (p = 4,2×10-10), ассоциированные с процентным содержанием жира в молоке (3). Исследования по GWAS-анализу среди разных популяций молочного скота США, Германии, Голландии, Австралии и Китая выявили пул общих генов, ответственных за показатели молочной продуктивности коров, в частности количество молочного жира (DGAT1, SCD1, GHR, EPS8, GPAT4), а также генов казеинового кластера (Hapmap24184-BTC-070077) (4-7). Отметим, что в популяции голштинского скота китайского происхождения по DGAT1 наблюдался эффект плейотропного действия гена по проценту и выходу молочного белка, удою (8).
Также ведутся работы по использованию в GWAS абсолютных фенотипических значений, или так называемых прямых фенотипов. При мета-анализе комплекса признаков молочной продуктивности, фертильности и типа телосложения молочного скота (9), а также оценке эффективности использования корма у мясного скота (10) были получены высокодостоверные значения ассоциаций с главными генами, что свидетельствует об их связи с фенотипической изменчивостью по этим показателям. Аналогичные исследования на свиньях пород крупная белая, ландрас и дюрок демонстрируют умеренную сходимость в результатах поиска QTL у животных, принадлежащих к разным популяциям (11-13). Интересно, что главным фактором мощности GWAS в этом случае выступает точность фенотипирования признаков и строгий учет факторов среды для отдельно взятого эксперимента. Практика использования данных многолетних наблюдений характерна в большей степени для исследовательских стад Европы, Северной Америки и Австралии и получения «чистых» фенотипов животных не столько по стандартным признакам продуктивности, сколько для показателей, требующих трудоемкого учета (продукция метана, остаточное потребление корма, жирно- и аминокислотный состав молока и мяса и др.) (14).
Еще одним эффективным инструментом геномного анализа служит оценка индивидуальной аутозиготности животных, или протяженных гомозиготных нуклеотидных фрагментов — паттернов ROH (runs of homo-zygosity), указывающих на использование близкородственного разведения. На примере анализа демографических событий в популяциях скота пинцгау, бурой швицкой и тирольской пород определены пороги по категориям длин ROH на основе вероятности наследования сегментов генома от общего предка 50, 25, шесть и три поколения назад (при ROH соответственно > 1, > 2, > 8 и > 16 Mb) (15). Закрепление в поколениях животных ROH с низкой частотой рекомбинации локусов может указывать на образование кластеров генов, находящихся под селекционным давлением, в первую очередь положительного отбора по признакам продуктивности, а также адаптации к изменению средовых условий (16, 17). Тем не менее ROH паттерны позволяют более эффективно, чем по родословной, оценивать степень геномного инбридинга (FROH) как в популяции (стаде), так и индивидуально для животного. Например, средняя вариабельность значений FROH между четырьмя стадами черно-пестрого скота Ленинградской области составляла 5,5-8,0 % для выборок животных от 46 до 85 гол. (18).
Описанные выше результаты GWAS и ROH анализов в большей степени относятся к изучению механизмов изменчивости генетической архитектуры количественных и качественных признаков у животных. Практическое направление использования геномных данных сосредоточено на разработке методов раннего отбора лучших генотипов — геномной селекции. В мире такой подход считается рутинной процедурой, в России — находится на этапе разработки и внедрения. Первые результаты показывают, что для молодых быков российского происхождения, используемых в воспроизводстве стад, значения геномного прогноза должны составлять не ниже +900 кг по молоку, +31 кг по молочному жиру и +23 кг по молочному белку (19).
Научная новизна настоящей экспериментальной работы заключается в интеграции методов геномного анализа и прогнозирования для животных трех поколений, находящихся одномоментно в высокопродуктивном стаде, с целью разработки модели их раннего отбора по комплексу показателей — степени геномного инбридинга и геномной племенной ценности. Полученные результаты подтвердили наше предположение о возможности использовать прямые фенотипические данные о продуктивности коров при поиске ассоциаций этих признаков с нуклеотидными мутациями и определении локусов в геноме, находящихся под давлением отбора. Впервые показано, что расширение референтной российской группы быков-производителей и коров может быть обеспечено за счет включения популяции голштинизированного черно-пестрого скота Урала.
Целью комплексных исследований был поиск полногеномных ассоциаций, а также локусов, сопряженных с интенсивностью отбора в популяции голштинизированного черно-пестрого скота Урала, для характеристики геномной вариабельности признаков молочной продуктивности и фертильности животных, принадлежащих к разным генерациям потомков.
Методика. Для процедуры генотипирования животных был использован биочип Bovine GGP 150K («Illumina/Neogen», США) с плотностью покрытия 138974 SNP. По результатам контроля качества чтения геномной информации и ведения племенного учета были выбраны генотипы с наличием не менее 110448 SNP для 85 гол., включая 9 быков-производителей голштинской породы (ОАО «Уралплемцентр», г. Екатеринбург, Свердловская обл.) и их потомков — 76 голштинизированных черно-пестрых коров и телок (ПАО «Каменское», Свердловская обл.). Средняя доля голштинских генов в выборке черно-пестрой породы составляла 97,7 % с колебаниями от 75 до 99 %. Для изучения динамики накопления гомозиготности и геномной племенной ценности в поколениях были сформированы экспериментальные группы животных и отобраны особи в трех генерациях родоначальниц и их потомков: матери матерей (25 гол., 2012-2014 годы рождения), матери (30 гол., 2015-2017 годы рождения) и дочери (21 гол., 2018-2019 годы рождения). Показатели молочной продуктивности в группах животных «мать матери» и «мать» составляли в среднем за 305 сут лактации соответственно по удою 7666 и 8461 кг молока, по массовой доле жира (МДЖ) 3,95 и 3,99 %, белка (МДБ) — 3,20 и 3,21 %, по молочному жиру (МЖ) 302,0 и 338,1 кг и по молочному белку (МБ) 245,0 и 274,4 кг.
Редактирование данных биочипов для построения файлов адаптированного расширения (.ped, .map, .fam, .bed, .bim), а также расчет полногеномных ассоциаций проводили в программе Plink 1.9 (20). Диаграммы распределения GWAS и ROH на хромосомах строили с помощью пакетов qqman и ggplot2 в среде программирования и визуализации языка R. Для GWAS анализа использовали усредненные показатели прямых фенотипов 44 коров за 1-5-ю законченные лактации. С помощью многофакторного дисперсионного анализа, реализованного в программе STATISTICA 10 («StatSoft, Inc.», США), изучали влияние факторов (год отела, ферма, бык-отец) на изменчивость фенотипических признаков у животных. На основе дизайна биочипа (https://genomics.neogen.com/pdf/slicks/ggp_bovine150k.pdf) предварительно отобрали селекционно значимые точечные мутации, картируемые в следующих генах и имеющие полиморфные варианты генотипов: DGAT1 (ген диацилглицерол-О-ацилтрансферазы 1), LEP (лептин), kCSN_A(CE) (k-казеин, аллели А, С, Е), kCSN_AB (k-казеин, аллели А, В), HH1, HH3 (гаплотипы фертильности), GHR (рецептор гена гормона роста); CSN2_I (b-казеин, аллельный вариант I); ABCG2 (АТФ-связывающая кассета подсемейства G); BLG (b-лактоглобулин); YellowFat (отвечает за изменения содержания каротина в жировых клетках).
ROH паттерны изучали с помощью программы cgaTOH (21) в объединенной выборке коров (матери матерей, матери), телок (дочери) и быков-отцов для расчета коэффициента геномного инбридинга в каждой из групп. Были приняты условия согласно подходу M. Ferenčaković (15), который предусматривает дифференциацию ROH на группы по их длине в связи с моментом возникновения демографического события: [1;2], (2;4], (4;8], (8;16] и > 16 Mb. Далее результаты визуализировали и определяли общие сегменты ROH, встречающиеся с наибольшей частотой не менее чем у 30-40 % животных (порог снижали до 30 % из-за небольшого размера экспериментальной выборки).
Аннотацию выявленных ассоциаций между SNP и фенотипами животных, а также локализацию ROH паттернов и входящих в них генов проводили по сборке генома крупного рогатого скота Bos_taurus_UMD_3.1.1 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000003055.6, дата обращения 30.01.2020 года). Для поиска QTL и регионов под давлением отбора на хромосомах, имеющих сопряженность с функциональными характеристиками животных, использовали базу данных CattleQTLdb (22).
Расчет геномных оценок племенной ценности (direct genomic values, DGV) по признакам молочной продуктивности в выборке из маточного поголовья трех генераций и быков-производителей осуществляли на основе российской референтной группы животных (591 гол.) (19) в соответствии с алгоритмом GBLUP, предложенным P.M. VanRaden (23).
Результаты. В задачи нашего исследования входило генотипирование быков-производителей голштинской породы и голштинизированных коров и телок черно-пестрой породы в поколениях мать матери—мать—дочь по 138 тыс. SNP. В соответствии с дизайном биочпа также планировалось определить носительство ценных аллелей генов, сопряженных с качественным и количественным составом молока, а также с рецессивными мутациями, вызывающими потерю репродуктивной способности. Кроме того, мы использовали данные GWAS и сопоставили их с прямыми показателями продуктивности и воспроизводительными качествами коров с целью поиска локусов количественных признаков в геноме крупного рогатого скота в уральской популяции. Наряду с этим была выполнена оценка степени геномного инбридинга в генерациях по группам мать матери—мать—дочь и быки-отцы коров и телок для уточнения использования информации в анализе результатов подбора родительских пар, а также поиске регионов в геноме (ROH), подверженных давлению отбора. Были рассчитаны значения DGV для признаков молочной продуктивности, что позволило изучить динамику их изменения в разных поколениях потомков.
На первом этапе мы определили частоты встречаемости генов, сопряженных с составом молока и фертильностью животных, степень полиморфизма и генетическое равновесие по этим генам в изученной уральской популяции голштинского скота (табл. 1).
Установлено, что по всем детектированным точечным мутациям в генах имелись полиморфные варианты генотипов с вариацией степени гомозиготности (по Робертсону) от 0,501 до 0,989. Мы обнаружили небольшое отклонение от генетического равновесия для гена k-казеина аллельного варианта А(СЕ) — χ2 = 3,894 (p < 0,05) при пороге χ2 = 3,840 (до этого значения равновесие не нарушается). Для гаплотипов фертильности HH1 и HH3 было выявлено носительство рецессивных мутаций, находящихся в гетерозиготном состоянии, с частотами встречаемости соответственно 1,0 и 2,1 %.
Желательные генотипы (включая гетерозиготы) по генам белков молока для κ-форм казеинов составляли по AE генотипу гена kCasein_A(CE) 20,8 %, по ВВ генотипу гена kCasein_AB 7,3 %, тогда как для β-формы — только 1,0 % (А1I генотип гена CSN2_I), что в целом специфично для популяции голштинского скота как более ориентированной на производство значительных объемов цельного молока, чем количества продукции его переработки (больший выход сыра, творога, масла). Для генов DGAT1 и ABCG2, сопряженных с соответственно процентным выходом жира и количеством молочного белка, получили умеренное распределение частот желательных генотипов по альтернативному аллелю 22 (AA) — 13,7 и 19,8 %, что указывает на наследственно предопределенные аспекты селекции по признакам количественного состава компонентов молока (частоты желательных аллелей для каждого из генов составляли соответственно 0,374 и 0,453). По гену β-лактоглобулина (BLG), связанного с аллергенностью молока коров для человека, частота встречаемости наиболее желательного генотипа AA, условно обозначаемого как 11, составляла в исследуемой популяции 9,5 % (31,1 % для аллеля 1), что свидетельствует о возможности отбора животных со специфичным статусом по указанному гену при создании стад для производства молока с низкими аллергенными свойствами.
Наряду с показателями молочной продуктивности важное значение имеет интенсивность роста и развития животных, способность к интенсивному откорму (у бычков). Согласно полученным нами данным, для генотипов скота по гену лептина и рецептору гена гормона роста желательные аллельные варианты составили соответственно 20,0 % (LEP_TT) и 64,6 % (GHR_AA).
С целью подтверждения гипотезы о геномной обусловленности показателей собственной продуктивности (так называемых прямых фенотипов) для признаков молочной продуктивности и фертильности коров провели GWAS анализ. Разделение на группы по поколениям матери матерей, матери и дочери проводилось для учета эффекта влияния популяционной структуры при ограниченной совокупности наблюдений, а также для выявления сегментов в геноме, имеющих общую идентичность по происхождению. Увеличение средней продуктивности за 1-5-ю лактации в поколении составило +795 кг по молоку, +0,04 % по МДЖ, +0,01 по МДБ, +36,1 кг по МЖ и +29,4 кг по МБ. Многофакторный дисперсионный анализ для определения влияния паратипических и генетических факторов на изменчивость признаков показал, что год отела, ферма и бык-отец не имели достоверной значимости (исключение — показатель МДБ, уровни значимости от p < 0,05 до p < 0,001). На этом основании далее при GWAS анализе использовали прямые фенотипы коров без корректировки на различия факторов среды.
Ни рисунке 1 представлены результаты анализа полногеномных ассоциаций с признаками молочной продуктивности и фертильности коров исследуемой выборки. Мы выявили значимую сопряженность между собственной продуктивностью животных по удою за 305 сут лактации и генотипом по SNP маркерам, локализованным на хромосомах крупного рогатого скота (BTA) BTA7, BTA10, BTA14, BTA16; по МДЖ и SNP маркерам на BTA1, BTA8, BTA11, BTA20; по МДБ и SNP маркерам BTA2, BTA 5, BTA7, BTA12, BTA16; по числу осеменений (ЧО) и SNP маркерам на BTA1, BTA7, BTA8, BTA21, BTA26.
На основе общедоступной базы данных CattleQTLdb были определены локусы количественных признаков в геноме скота, в которые входят гены и SNP, достоверно ассоциированные с селекционными показателями в изученной популяции скота Урала (табл. 2). Для удоя были детектированы значимые полиморфизмы в генах ZNF16, ARHGAP39, EFR3A, KCNQ3, KHDRBS3, ассоциированные с удоем, выходом продукции молочного жира и белка, которые соответствовали локусам, найденным в аналогичных работах зарубежных авторов (24). Для генов HCN4 и PCSK1 была характерна сопряженность с параметрами роста и развития, а также поведенческими реакциями, что позволяет говорить об их возможном влиянии на молочную продуктивность (25, 26).
Для величины удоя на BTA14 нами выявлен локус в позиции 1,44-1,59 Mb, в который входили три наиболее значимых гена и от которого на расстоянии 210 kb находится ген DGAT1, играющий важную роль в превращении диацилглицеридов в триацилглицериды с участием ацил-КоА сложных эфиров жирных кислот при синтезе жиров в организме (табл. 2). Ассоциации с массовой долей жира были средне достоверными (p-value 1,21×10-6-2,79×10-6), при этом связь с QTL показали гены ANTXR1, ZBTB38, PRUNE2, MYO10, аннотированные в большей степени признаками темперамента и остаточного потребления корма, а также параметрами роста, длины туловища и обхвата груди (27-30).
По массовой доле белка при GWAS анализе проявились более сильные ассоциации (p-value 4,38×10-6-1,08×10-7). Отметим, что для генов COL3A1, IGF1 и PAHполученные нами данные не полностью совпали с имеющимися сообщениями о связи картируемых в этих генах QTL с показателями мраморности туши, удоем, процентом и выходом молочного белка, живой массой, числом осеменений, а также с рядом параметров оценки качества вымени и ног (31-33).
Ассоциации QTL с числом осеменений (в расчете на одно плодотворное) были обусловлены функциональной связью этого признака с генами, контролирующими репродуктивные качества животных. В частности, для QTL, которые локализованы в генах, отвечающих за овариальную регуляцию и атрезию фолликулов(ген SERPINA5, включая близлежащие варианты суперсемейства SERPINA1, SERPINA10, SERPINA3-2, SERPINA11) (34), уровень значимости ассоциаций составил p = 4,13×10-7; за живую массу в возрасте 18 мес в период плодотворного осеменения (ген ARHGAP31) (35) — p = 1,79×10-6; за раннее преимплантационное развитие эмбриона (ген CHD1)(36) — p = 3,17×10-6; за оплодотворяемость дочерей быков (ген STXBP5L)(24) — p = 1,14×10-5. Мы также обнаружили полиморфизмы в гене LOXL2 (24), с которым связывают число осеменений и выход молочного жира (р = 1,18×10-5), и в гене FBP2 (37), обусловливающем параметры роста у скота молочных пород (р = 1,70×10-5). Однако следует отметить, что также были выявлены ассоциации (p = 8,92×10-6) с полиморфизмами в генах, не связанных с репродуктивными качествами, например в генах резистентности к ряду паразитарных заболеваний (кластер длиной 22,1 kb на BTA16, включающий KIF14, DDX59 и CAMSAP2) (38).
Обнаруженные в результате GWAS анализа однонуклеотидные замены в значительной степени соответствовали генам и QTL, ранее аннотированным другими исследователями для признаков молочной продуктивности и фертильности крупного рогатого скота. Однако по некоторым SNP сопряженность с прямыми фенотипами животных подтверждалась только косвенно, что указывает либо на плейотропное действие этих SNP, либо на иную природу комбинативной изменчивости учитываемых количественных показателей (точность фенотипирования, дрейф генов, интенсивность отбора или его полное отсутствие).
В настоящее время развитие популяции голштинского скота в том или ином регионе разведения сопряжено с определенными трудностями — необходимостью избегать близкородственных связей, контролировать степень гомозиготности в популяции и распространение летальных рецессивных мутациями. Кроме того, требуется воспроизводство собственных племенных ресурсов (быков-производителей, ремонтного молодняка) с заданной племенной ценностью. Достижение высоких показателей продуктивности у коров возможно за счет создания генотипа, устойчиво передающего ценные аллели и/или гаплоблоки из поколения в поколение при умеренном уровне гомозиготности.
Алгоритм поиска ROH паттернов предназначен для сканирования участков последовательно расположенных по всей длине хромосомы гомозиготных однонуклеотидных замен. ROH определяли при условии, что 15 или более последовательных гомозиготных SNP присутствовали на исследуемом участке генома при плотности не менее 1 SNP на каждые 100 kb с промежутками между ними не более 1000 kb. Также были приняты следующие условия: начиная с группы ROH длиной (8;16] Mb, допускалось наличие двух пропущенных SNP на сегмент, при > 16 Mb — четырех пропущенных нуклеотидных замен и одного гетерозиготного аллеля. В таблице 3 приведены данные по числу SNP, входящих в ROH разной длины, с учетом принятых допущений.
С увеличением длины паттернов число SNP, входивших в сегмент, возрастало практически пропорционально до максимума (2214 маркеров). Средняя длина ROH с увеличением паттерна от 1-2 Mb до > 16 Mb возрастала от 1,40 до 22,01 Mb, в то время как общая длина ROH (сумма всех сегментов в среднем на животное по градациям) значительно не менялась и даже имела тенденцию к снижению — от 48,3 Mb до 42,7 Mb.
Поиск каузальных мутаций в генах с помощью визуализации данных (рис. 2) и анализа частоты встречаемости ROH паттернов в изучаемой популяции молочного скота (табл. 4) позволил установить ряд консервативных регионов в геноме (кластеров с входящими в них протеин-кодирующими генами), которые, как мы предполагаем, находятся под давлением отбора.
В связи с ограниченностью размера экспериментальной выборки на-чало и конец каждого детектированного ROH региона приведено как среднее значение в Mb со стандартным отклонением (±s) (см. табл. 4), чтобы максимально точно указать размах вариации для гомозиготного сегмента. Проанализировав распределение ROH по 29 хромосомам в трех поколениях животных, мы выявили пять геномных регионов с максимальной встречаемостью паттернов не менее чем у 30 % особей: на BTA12 — участок 35,3-39,7 Mb, на BTA14 — участки 1,6-5,1 Mb и 24,4-26,6 Mb, на BTA26 — 22,1-23,9 Mb, на BTA29 — 38,0-42,0 Mb. Аннотирование находящихся в этих регионах SNP, выполненное для поиска наиболее значимых генов и формируемых ими QTL, показало, что от 35 до 73 % особей в общей выборки имели аналогичное расположение ROH паттернов с детектируемыми позициями.
После визуального поиска и анализа частоты встречаемости ROH была проведена аннотация генов, формирующих кластеры повышенного селекционного давления. Среди 25 генов на BTA12 к наиболее значимым отнесли только два — IL17D (интeрлейкин 17Д) (39) и PARP4, кодирующий поли(АДФ-рибоза)-полимеразу (40). Эти гены вовлечены в иммунные процессы защиты молочной железы, а также адипогенез. В целом на основе функциональной аннотации в базе данных Cattle QTL Database (https://www.animalgenome.org/cgi-bin/QTLdb/BT/index) можно резюмировать, что в указанном регионе BTA12 сосредоточены QTL, сопряженные с выходом молочного жира и белка, количеством соматических клеток, продолжительностью продуктивной жизни, персистентностью лактации, живой массой, трудностью отела, мертворождениями, числом осеменений на одно оплодотворение, качеством вымени и конечностей.
Для BTA14 по ROH выделились два региона, которые достаточно информативны с точки зрения селекции молочного скота. В первый вошли 66 генов, из которых наиболее изучены DGAT1 (ген диацилглицерол-ацилтрансферазы 1), PLEC (плектин) и GRINA (глутаминовый рецептор), вовлеченные в процессы синтеза липидных компонентов — жира молока и некоторых жирных кислот (3). У животных QTL в большей степени были ассоциированы с удоем за лактацию, содержанием жира, казеина, фосфора, кальция, жирных кислот молока, количеством соматических клеток, предрасположенностью к маститам, кетозу, с устойчивостью к туберкулезу и тепловому стрессу. Второй регион на хромосоме содержал 18 генов, которые входили в QTL, ответственные за параметры роста и развития животных, живую массу, среднесуточный прирост, качественные и количественные послеубойные характеристики туш, участие в синтезе инсулиноподобного фактора роста 1. Одним из определяющих в этом случае служит ген плейоморфной аденомы 1 PLAG1 (он отвечает за параметры роста и репродукцию у животных, кроме того, продукт этого гена выступает в роли фактора транскрипции, регулирующего экспрессию инсулиноподобных факторов роста) (41).
На BTA26 мы детектировали 18 генов. При этом продукты генов SCD (ген стеароил-КоА-десатуразы 1), PKD2L1,BTRC,ELOVL3,GBF1,SUFU и CNNM2 — это непосредственные предшественники в синтезе жирных кислот молока (линоленовой, стеариновой, мирист- и пальмитолеиновой, мононенасыщенных и ряда других) (42). Кроме того, на BTA26 мы обнаружили QTL, связанные с удоем, содержанием жира, белка, лактозы, линолевой, лауриновой, миристиновой и конъюгированной линолевой жирных кислот, казеина молока, а также с качественными характеристиками вымени коров (42).
Обнаружение на BTA29 генов, входящих в гомозиготный локус, позволяет нам сделать заключение об их влиянии на фертильность животных. К таким генам относятся PAG1-PAG20, которые кодируют группу гликопротеинов, ассоциированных со стельностью, а также гены INCENP и SLC3A2, для которых на основании анализа базы данных по QTL показана связь с подвижностью сперматозоидов и общим индексом фертильности (43, 44). В свою очередь, в ROH паттерне нами был также выявлен кластер генов десатуразы жирной кислоты (FADS1-3) — ключевого энзима в метаболизме жирных кислот молока у коров (45). Кроме того, указанный QTL регион также обусловливал изменчивость по таким признакам, как интервал между отелом и первой охотой (эструсом), число плодотворных осеменений, качество семени, фертильность дочерей быка, содержание белка, казеина, жира в молоке.
Применение паттернов гомозиготности для поиска «отпечатков» селекции в геноме животных этим не исчерпывается. Используя значение суммы общей протяженности ROH на особь, деленную на общую длину аутосомного генома (для настоящего исследования — 2516398000 bp), можно оценить индивидуальную аутозиготность особи. Предполагается, что по числу и сумме протяженных сегментов ROH можно судить об уровне так называемого геномного инбридинга, который должен показать более высокую точность по сравнению с родословной.
В этой связи мы рассчитали степень инбридинга на основе протяженности ROH в поколениях мать матери—мать—дочь и в контрольной группе отцов коров (табл. 5). Это обеспечило более точные результаты анализа накопления гомозиготности в популяции при последовательном подборе родительских пар животных. Наиболее наглядную оценку геномного инбридинга мы получили по градации FROH > 1 Mb: был выявлен достоверный рост в поколениях мать матери—мать от 8,6 до 9,8 %, или на 1,2 % (p < 0,05), и мать—дочь — от 9,8 до 12,7 %, или на 2,9 % (p < 0,001). Показатель FROH > 1 Mb у быков-отцов достигал значения 13,5 %. Существует мнение, что использование ROH > 1 Mb ведет к завышению показателя геномного инбридинга из-за затруднений при выявлении общих предков в родословной и высокой частоты рекомбинации, поэтому ряд авторов считают целесообразным определять FROH по сегментам свыше 4 Mb (18). Таким образом, популяция голштинизированного черно-пестрого скота (на примере высокопродуктивного уральского стада) показывает накопление общей гомозиготности и, как следствие, формирование группы, консолидированной по генетическим особенностям, присущим голштинской породе. При увеличении размера ROH паттернов более чем до 16 Mb наблюдаемое значение инбридинга в поколениях животных было ниже и составляло соответственно 1,4; 1,4 и 2,6 % (p < 0,001). Это свидетельствует о том, что значимую долю в оценке величины гомозиготности дают ROH с относительно небольшой протяженностью, которая отражает демографические события на породном уровне в целом. Использование ограниченного числа производителей-основателей в современной популяции скота (генеалогических комплексов — линий и ветвей породы), селекционированных на получение высокой молочной продуктивности, привело к повышению общей степени гомозиготности. Дальнейший рост геномного инбридинга (свыше 15,0 %) должен быть контролируемым для снижения возможного отрицательного влияния на репродуктивные признаки животных.
Предварительный анализ фенотипических показателей коров в сопоставлении с величиной их геномного инбридинга указывал на положительную корреляцию между этими параметрами: по удою — r = 0,464 (p < 0,01); по МДЖ — r = 0,121, МЖ — r = 0,463 (p < 0,01); МБ — r = 0,389 (p < 0,05); ЧО — r = 0,168 (за исключением МДБ с r = -0,400 (p < 0,01). То есть с ростом общей гомозиготности (ROH) одновременно улучшались показатели по удою, выходу молочного жира и белка при умеренном снижении показателей фертильности.
На основе российской референтной группы молочного скота мы рассчитали значения DGV для двух генераций коров, телок, а также для быков-отцов (см. табл. 5). Показано, что матери достоверно превосходили поколение матерей матерей в отношении геномной племенной ценности по удою (+157 кг молока, p < 0,01), МЖ (+6,6 кг, p < 0,01) и МБ (+4,1 кг, p < 0,05), уступая при этом по МДЖ (на -0,003 %, недостоверно) и МДБ (-0,013 %, p < 0,05). В генерации дочерей наблюдалось положительная динамика роста DGV по сравнению с матерями (на +32 кг по удою молока, +2,2 кг по МЖ, +1,8 кг по МБ, +0,007 % по МДЖ и +0,008 % по МДБ). Быки — отцы изучаемого потомства показали наибольшую геномную племенную ценность, однако, как и их дочери, не имели достоверного превосходства над генераций матерей в экспериментальной выборке. Это может свидетельствовать о выходе на селекционное плато в стаде вследствие насыщения генотипа потомков сочетанием ценных аллелей для признаков молочной продуктивности в результате подбора родительских пар. Дальнейшее генетическое улучшение особей возможно за счет использования новых генераций быков-производителей с бóльшими значениями DGV либо с высокой племенной ценностью, более точно определенной по качеству потомства.
Итак, выполненное исследование показало, что контроль моногенных генетических мутаций (дефектов), а также селекционно обусловленных ДНК маркеров может позволить более эффективно отбирать животных на уровне стада (или популяции в целом) и использовать их в подборе родительских пар для получения потомства с заданными племенными качествами. Анализ полногеномных ассоциаций с показателями молочной продуктивности и фертильности коров позволил установить каузальные мутации, локализованные близко к генам либо внутри них, из которых наиболее значимы нуклеотидные замещения в генах ZNF16, ARHGAP39, EFR3A, KCNQ3, KHDRBS3,IGF1, SERPINA5, ARHGAP31,CHD1 иSTXBP5L. Достоверность значимых ассоциаций по точечным мутациям находилась в пределах p < 2,3×10-5-1,1×10-7. Эти генетические полиморфизмы согласуются с описанными для российского голштинизированного черно-пестрого скота, а также для голштинской породы в Северной Америке и ряде европейских стран. На основе ROH анализа установлены геномные регионы, подверженные наибольшему селекционному давлению. Они расположены на 12-й, 14-й, 26-й и 29-й хромосомах и содержат QTL, сопряженные с признаками молочной продуктивности, фертильности животных, жирнокислотным составом молока коров, параметрами экстерьера, роста и развития. На основе паттернов гомозиготности уточнены показатели степени инбридинга у изученной группы скота. Это пример подхода, который может использоваться в программах разведения для подбора животных на уровне отдельных стад и популяции в целом. Анализ геномного инбридинга показал положительную динамику в поколениях, при этом одновременно повышалась прогнозируемая величина геномной племенной ценности животных. Эти данные подтверждают тот факт, что комплекс инструментов геномного анализа позволяет эффективнее планировать получение особей, которые бы отвечали экономическим потребностям молочного скотоводства. Пополнение российской референтной группы молочного скота за счет включения в нее региональных популяций (как генотипированных коров, так и быков-производителей) создает перспективы для расширения прогноза геномной племенной ценности при воспроизводстве как бычьего, так и маточного поголовья голштинизированного черно-пестрого и голштинского скота.
1. Оценка частоты встречаемости селекционно значимых генетических маркеров в изученной популяции крупного рогатого скота голштинизированной черно-пестрой породы (n = 85, ПАО «Каменское», Свердловская обл., 2019 год) |
||||||||
Ген |
Частота |
Генотип |
Частота аллелей |
χ2 |
Ca |
|||
11 |
12 |
22 |
1 |
2 |
||||
DGAT1 |
Н |
0,389±0,035 |
0,474±0,036 |
0,137±0,025 |
0,626 |
0,374 |
0,014 |
0,532 |
О |
0,392 |
0,468 |
0,140 |
|||||
LEP |
Н |
0,200±0,021 |
0,547±0,026 |
0,253±0,022 |
0,474 |
0,526 |
1,816 |
0,501 |
О |
0,224 |
0,499 |
0,277 |
|||||
kCasein_A(CE) |
Н |
0,792±0,017 |
0,208±0,017 |
0 |
0,896 |
0,104 |
3,894 |
0,813 |
О |
0,803 |
0,187 |
0,010 |
|||||
kCasein_AB |
Н |
0,531±0,021 |
0,396±0,020 |
0,073±0,011 |
0,729 |
0,271 |
0,734 |
0,605 |
О |
0,532 |
0,395 |
0,073 |
|||||
HH3 |
Н |
0,990±0,004 |
0,010±0,004 |
0 |
0,995 |
0,005 |
0,008 |
0,989 |
О |
0,990 |
0,010 |
0 |
|||||
HH1 |
Н |
0,979±0,006 |
0,021±0,006 |
0 |
0,010 |
0,990 |
0,032 |
0,979 |
О |
0,979 |
0,021 |
0 |
|||||
GHR |
Н |
0,646±0,035 |
0,333±0,034 |
0,021±0,010 |
0,813 |
0,187 |
0,849 |
0,695 |
О |
0,660 |
0,305 |
0,035 |
|||||
CSN2_I |
Н |
0,990±0,005 |
0,010±0,005 |
0 |
0,995 |
0,005 |
0,005 |
0,989 |
О |
0,990 |
0,010 |
0 |
|||||
ABCG2 |
Н |
0,292±0,033 |
0,510±0,036 |
0,198±0,029 |
0,547 |
0,453 |
0,085 |
0,504 |
О |
0,299 |
0,496 |
0,205 |
|||||
BLG |
Н |
0,095±0,012 |
0,432±0,021 |
0,474±0,021 |
0,311 |
0,698 |
0,018 |
0,572 |
О |
0,096 |
0,428 |
0,475 |
|||||
YellowFat |
Н |
0,990±0,004 |
0,010±0,004 |
0 |
0,005 |
0,995 |
0,011 |
0,989 |
О |
0,990 |
0,010 |
0 |
|||||
Примечание. Для генотипов и частоты аллелей приведены варианты кодирования. HH1 и HH3 — наименования указаны в соответствии с дизайном чипа.Ca — коэффициент гомозиготности по Робертсону. Н и О — соответственно наблюдаемая и ожидаемая частота. |
||||||||
2. Значимые однонуклеотидные мутации, ассоциированные с фенотипическими признаками коров в изученной группе животных голштинизированной черно-пестрой породы (ПАО «Каменское», Свердловская обл., 2019 год) |
|||||
SNP |
BTA |
Позиция, bp |
p-value |
Ближайший ген |
Расстояние до гена, bp |
Средний удой за 305 сут лактации, кг |
|||||
BovineHD1600003292 |
16-я |
12318853 |
4,18×10-6 |
CDC73 |
+325765 |
BovineHD1400000152 |
14-я |
1439476 |
8,54×10-6 |
ZNF16 |
Внутри |
BovineHD1400000187 |
14-я |
1585385 |
8,54×10-6 |
ARHGAP39 |
Внутри |
BTA-34956-no-rs |
14-я |
1514056 |
8,54×10-6 |
ZNF7 |
+1864 |
BovineHD1000006525 |
10-я |
19972763 |
1,58×10-5 |
HCN4 |
+7449 |
BovineHD1400002920 |
14-я |
10319796 |
1,58×10-5 |
EFR3A |
-37385 |
ARS-BFGL-NGS-42106 |
7-я |
97772240 |
2,23×10-5 |
ELL2 |
-78731 |
BovineHD0700028539 |
7-я |
97833516 |
2,23×10-5 |
PCSK1 |
+322158 |
BovineHD4100010843 |
14-я |
7332355 |
2,31×10-5 |
KHDRBS3 |
+99747 |
Средняя массовая доля жира, % |
|||||
BovineHD1100019146 |
11-я |
67734294 |
1,21×10-6 |
GFPT1 |
Внутри |
Hapmap59899-ss46527105 |
11-я |
67678534 |
1,21×10-6 |
ANTXR1 |
-88003 |
ARS-BFGL-NGS-36975 |
1-я |
128799966 |
1,83×10-6 |
ZBTB38 |
-263595 |
BovineHD0100036417 |
1-я |
128823979 |
1,83×10-6 |
SPSB4 |
+6929 |
BovineHD0800015859 |
8-я |
52869025 |
1,83×10-6 |
RFK |
-65127 |
BovineHD0800015899 |
8-я |
52984674 |
1,83×10-6 |
PRUNE2 |
Внутри |
BTB-00347944 |
8-я |
53031744 |
1,83×10-6 |
PRUNE2 |
Внутри |
ARS-BFGL-NGS-38258 |
20-я |
56721394 |
2,79×10-6 |
MYO10 |
-149181 |
Средняя массовая доля белка, % |
|||||
ARS-BFGL-NGS-45195 |
16-я |
63612072 |
1,08×10-7 |
STX6 |
Внутри |
BovineHD4100009755 |
12-я |
81991589 |
3,23×10-7 |
ITGBL1 |
Внутри |
BTA-87771-no-rs |
12-я |
82056537 |
3,23×10-7 |
ITGBL1 |
Внутри |
BovineHD0200021465 |
2-я |
74934453 |
3,31×10-7 |
IGDCC3 |
+455733 |
BovineHD1200023614 |
12-я |
82094359 |
4,37×10-7 |
ITGBL1 |
Внутри |
ARS-BFGL-NGS-102876 |
16-я |
63521833 |
2,00×10-6 |
XPR1 |
-19726 |
BovineHD0200009386 |
2-я |
31672054 |
2,52×10-6 |
COBLL1 |
Внутри |
BovineHD0200002141 |
2-я |
7215096 |
2,95×10-6 |
COL3A1 |
+102191 |
Hapmap53461-rs29027660 |
7-я |
102855103 |
4,37×10-6 |
AP3S1 |
142023 |
BovineHD0500018676 |
5-я |
66809794 |
4,38×10-6 |
IGF1 |
-206095 |
Среднее число осеменений |
|||||
BovineHD2100017363 |
21-я |
59895529 |
4,13×10-7 |
SERPINA5 |
-99716 |
ARS-BFGL-NGS-119213 |
26-я |
47375257 |
4,87×10-7 |
DOCK1 |
-79054 |
BovineHD0100018320 |
1-я |
64773642 |
1,79×10-6 |
ARHGAP31 |
Внутри |
BovineHD0700029413 |
7-я |
100618564 |
3,17×10-6 |
CHD1 |
-51472 |
BovineHD1600023747 |
16-я |
81193491 |
8,92×10-6 |
KIF14 |
-1809 |
BovineHD1600023756 |
16-я |
81215628 |
8,92×10-6 |
CAMSAP2 |
+62155 |
BovineHD0100018663 |
1-я |
66151741 |
1,14×10-5 |
STXBP5L |
+61871 |
ARS-BFGL-NGS-108666 |
8-я |
71352779 |
1,18×10-5 |
LOXL2 |
Внутри |
BovineHD0800021457 |
8-я |
71309172 |
1,18×10-5 |
LOXL2 |
Внутри |
BovineHD0800024578 |
8-я |
82536035 |
1,70×10-5 |
FBP2 |
-97218 |
Примечание. SNP — однонуклеотиднные замены представлены в порядке снижения уровня достоверности ассоциаций (p-value), BTA — хромосома крупного рогатого скота; «+» — расстояние по направлению к гену от соответствующего SNP, «-» — расстояние против направления. |
|||||
3. Статистические параметры числа однонуклеотидных полиморфизмов (SNP) для паттернов гомозиготности (ROH) разной протяженности в изученной группе животных голштинизированной черно-пестрой породы (n = 85, ПАО «Каменское», Свердловская обл., 2019 год) |
|||||
Параметр |
Длина ROH по группам, Mb |
||||
[1;2] |
(2;4] |
(4;8] |
(8;16] |
> 16 |
|
Число SNP |
58,6±0,4 |
122,0±1,1 |
233,8±2,0 |
449,8±5,0 |
904,2±27,0 |
Стандартное отклонение |
22,3 |
44,4 |
67,8 |
110,2 |
268,3 |
Минимум |
15 |
15 |
27 |
253 |
617 |
Максимум |
289 |
476 |
668 |
1050 |
2214 |
Средняя длина ROH, Mb |
1,40±0,01 |
2,82±0,01 |
5,49±0,03 |
10,77±0,10 |
22,01±0,65 |
Общая длина ROH, Mb |
48,3±1,2 |
59,0±2,0 |
71,0±2,6 |
63,8±3,4 |
42,7±3,9 |
4. Геномные регионы и наиболее значимые протеин-кодирующие гены, находящиеся под давлением отбора и идентифицированные с помощью ROH (протяженные гомозиготные фрагменты) в изученной группе животных голштинизированной черно-пестрой породы (n = 76, ПАО «Каменское», Свердловская обл., 2019 год) |
|||||
BTA |
Средняя детектируемая |
Доля животных c ROH в указанном регионе, % |
Число |
Наиболее значимые гены |
|
начало±σ |
конец±σ |
||||
12-я |
35,258±1,967 |
39,747±3,407 |
44 |
25 |
IL17D, PARP4 |
14-я |
1,580±0,454 |
5,102±2,424 |
35 |
66 |
ARHGAP39, LRRC14, PPP1R16A, FOXH1, CYHR1, TONSL, CPSF1, ADCK5, SLC52A2, TMEM249, SCRT1, DGAT1, BOP1, MROH1, MAF1, OPLAH, SMPD5, SPATC1, PLEC, GRINA, MAPK15, EPPK1, SCRIB, EEF1D, RHPN1, GPIHBP1, GML, ADGRB1, TSNARE1, SLC45A4, DENND3, PTK2, AGO2, TRAPPC9, KCNK9 |
14-я |
24,412±1,828 |
26,621±2,346 |
59 |
18 |
XKR4, PLAG1, CHCHD7, SDR16C5- SDR16С6, FAM110B, SDCBP, NSMAF, TOX |
26-я |
22,142±1,102 |
23,877±1,878 |
73 |
48 |
PKD2L1, SCD, BTRC, ELOVL3, GBF1, SUFU, CNNM2 |
29-я |
37,953±1,177 |
41,982±2,334 |
62 |
117 |
PAG1-PAG20, FADS1-FADS3, INCENP, SLC3A2 |
Примечание. BTA — хромосома крупного рогатого скота. |
|||||
5. Сравнительные оценки уровня геномного инбридинга (FROH), общей протяженности гомозиготных фрагментов (ROHtotal, Mb) в связи с длиной ROH, поколением животных дочь—мать—мать матери и величиной непосредственной геномной племенной ценности (DGV) в изученной группе животных голштинизированной черно-пестрой породы (n = 85, M±SEM, ПАО «Каменское», Свердловская обл., 2019 год) |
||||
Показатель |
Поколение животных (годы рождения) |
|||
мать матери, n = 25 |
мать, n = 30 |
дочь, n = 21 |
отцы коров, n = 9 |
|
Уровень геномного инбридинга в изучаемой выборке (ROH) |
||||
FROH > 1 |
0,086±0,004 |
0,098±0,004* |
0,127±0,005*** |
0,135±0,009 |
FROH > 2 |
0,068±0,004 |
0,080±0,004* |
0,105±0,005*** |
0,114±0,009 |
FROH > 4 |
0,051±0,004 |
0,061±0,003* |
0,083±0,005*** |
0,092±0,009 |
FROH > 8 |
0,028±0,002 |
0,034±0,003 |
0,053±0,004*** |
0,059±0,009 |
FROH > 16 |
0,014±0,002 |
0,014±0,002 |
0,026±0,003*** |
0,032±0,005 |
ROHtotal > 1 |
215,3±10,1 |
247,3±9,3* |
319,9±12,7*** |
338,7±23,1 |
Геномная племенная ценность по признакам молочной |
||||
Удой, кг |
448,7±37,8 |
645,5±39,7** |
677,1±57,1 |
730,3±113,0 |
МДЖ, % |
0,0005±0,0108 |
-0,0024±0,0101 |
0,0049±0,0133 |
-0,0295±0,0126 |
МЖ, кг |
18,3±1,4 |
24,9±1,6** |
27,1±2,1 |
27,3±15 |
МДБ, % |
-0,0017±0,0032 |
-0,0144±0,0042* |
-0,0059±0,0041 |
-0,0165±0,0056 |
МБ, кг |
9,6±1,3 |
13,7±1,2* |
15,5±1,9 |
16,7±3,1 |
Примечание. МДЖ — массовая доля жира, МЖ — молочный жир, МДБ — массовая доля белка, МБ — молочный белок. |
||||
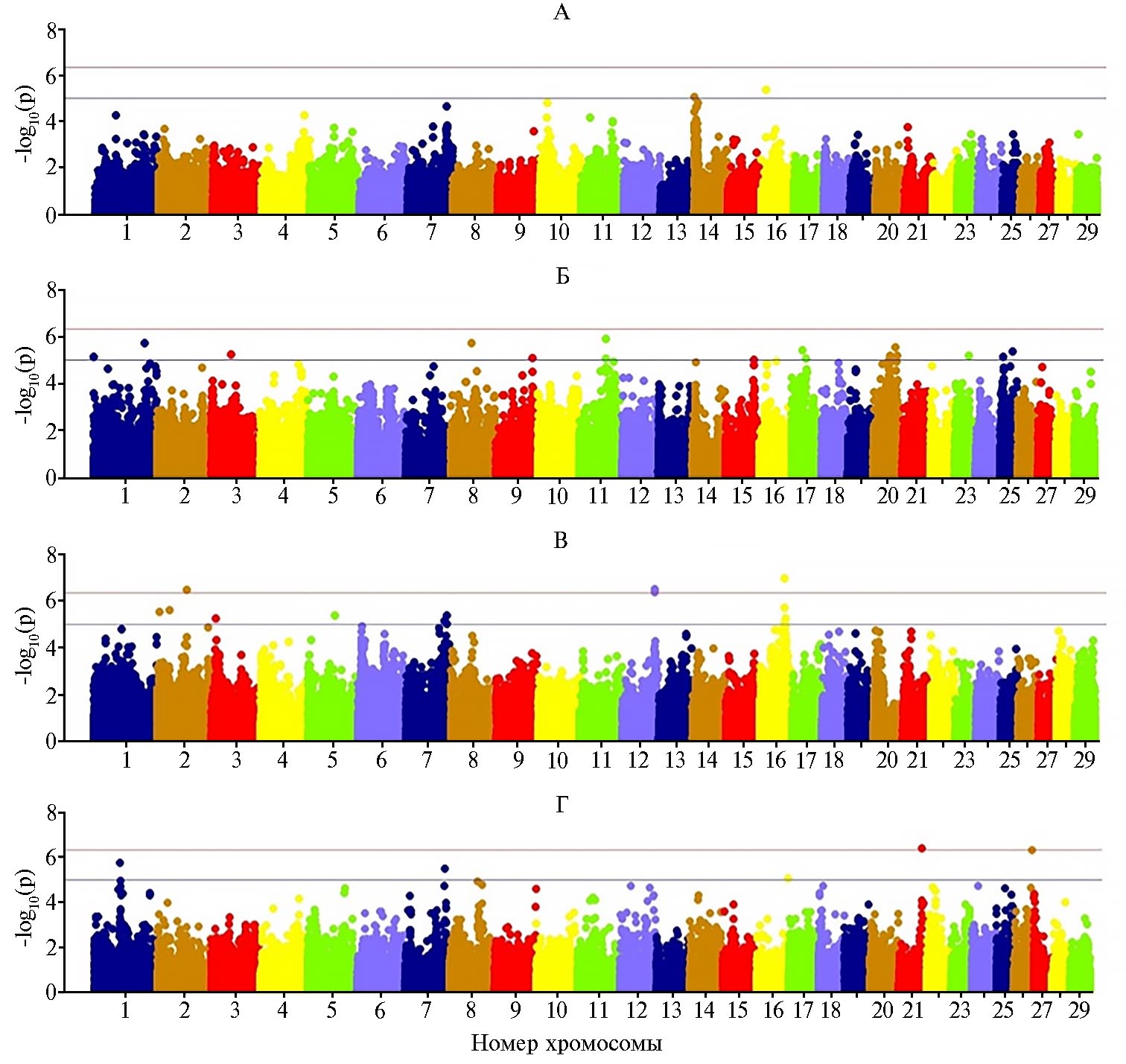
Рис. 1. Распределение однонуклеотидных мутаций по хромосомам в изученной группе коров голштинизированной черно-пестрой породы в связи с уровнем статистической значимости для показателей собственной продуктивности в среднем за 305 сут лактации: А — удой за лактацию, Б — массовая доля жира, В — массовая доля белка, Г — число осеменений. Верхняя горизонтальная линия — порог достоверности для полногеномных ассоциаций -log10(p) = 4,5×10-7, нижняя горизонтальная линия — порог достоверности для суггестивных ассоциаций -log10(p) = 1,0× 10-5 (n = 44, ПАО «Каменское», Свердловская обл., 2019 год).

Рис. 2. Идентификация ROH паттернов для селекционно значимых локусов (выделены прямоугольной рамкой) на 12-й (А), 14-й (Б), 26-й (В) и 29-й (Г) хромосомах крупного рогатого скота (ВТА) в изученной группе животных голштинизированной черно-пестрой породы (строка соответствует одному животному; n = 76, особи женского пола разных генераций, ПАО «Каменское», Свердловская обл., 2019 год).
ЛИТЕРАТУРА
- Зиновьева Н., Стрекозов Н., Янчуков И., Ермилов А., Ескин Г. Система геномной оценки скота: первые результаты. Животноводство России, 2015, 3: 27-30.
- Кудинов А.А., Масленникова Е.С., Племяшов К.В. Генетический прогресс ключевой аспект совершенствования молочного животноводства развитых стран. Зоотехния, 2019, 1: 2-6.
- Sermyagin А.А., Gladyr Е.А., Plemyashov K.V., Kudinov А.A., Dotsev A.V., Deniskova Т.Е., Zinovieva N.A. Genome-wide association studies for milk production traits in Russian population of Holstein and Black-and-White cattle. Proceedings of the Scientific-Practical Conference «Research and Development — 2016» /K. Anisimov et al. (eds.). Springer, Cham, 2018: 591-599 (doi: 10.1007/978-3-319-62870-7_62).
- Qanbari S., Pimentel E.C.G., Tetens J., Thaller G., Lichtner P., Sharifi A.R., Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Animal Genetics, 2010, 41(4): 377-389 (doi: 10.1111/j.1365-2052.2009.02016.x).
- Wang X., Wurmser C., Pausch H., Jung S., Reinhardt F., Tetens J., Thaller G., Fries R. Identification and dissection of four major QTL affecting milk fat content in the German Holstein-Friesian population. PLoS ONE, 2012, 7(7): e40711 (doi: 10.1371/journal.pone.0040711).
- Maurice-Van Eijndhoven M.H.T., Bovenhuis H., Veerkamp R.F., Calus M.P.L. Overlap in genomic variation associated with milk fat composition in Holstein Friesian and Dutch native dual-purpose breeds. Journal of Dairy Science, 2015, 98(9): 6510-6521 (doi: 10.3168/jds.2014-9196).
- Chamberlain A.J., Hayes B.J., Savin K., Bolormaa S., McPartlan H.C., Bowman P.J., Van Der Jagt C., MacEachern S., Goddard M.E. Validation of single nucleotide polymorphisms associated with milk production traits in dairy cattle. Journal of Dairy Science, 2012, 95(2): 864-875 (doi: 10.3168/jds.2010-3786).
- Fang M., Fu W., Jiang D., Zhang Q., Sun D., Ding X., Liu J. A multiple-SNP approach for genome-wide association study of milk production traits in Chinese Holstein cattle. PLoS ONE, 2014, 9(8): e99544 (doi: 10.1371/journal.pone.0099544).
- Xiang R., MacLeod I.M., Bolormaa S., Goddard M.E. Genome-wide comparative analyses of correlated and uncorrelated phenotypes identify major pleiotropic variants in dairy cattle. Scientific Reports, 2017, 7: 9248 (doi: 10.1038/s41598-017-09788-9).
- Zhang F., Wang Y., Mukiibi R., Chen L,, Vinsky M., Plastow G., Basarab J., Stothard P., Li G. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: feed efficiency and component traits. BMC Genomics, 2020, 21: 36 (doi: 10.1186/s12864-019-6362-1).
- Reyer H., Shirali M., Ponsuksili S., Murani E., Varley P.F., Jensen J., Wimmers K. Exploring the genetics of feed efficiency and feeding behaviour traits in a pig line highly selected for performance characteristics. Molecular Genetics and Genomics: MGG, 2017, 292(5): 1001-1011 (doi: 10.1007/s00438-017-1325-1).
- Мельникова Е.Е., Бардуков Н.В., Форнара М.С., Костюнина О.В., Сермягин А.А., Brem G., Зиновьева Н.А. Влияние генотипов по ДНК-маркерам на воспроизводительные качества свиней пород крупная белая и ландрас. Сельскохозяйственная биология, 2019, 54(2): 227-238 (doi: 10.15389/agrobiology.2019.2.227rus).
- Белоус А.А., Сермягин А.А., Костюнина О.В., Brem G., Зиновьева Н.А. Изучение генетической архитектуры конверсии корма у хряков (Sus scrofa) породы дюрок на основе полногеномного анализа SNP. Сельскохозяйственная биология, 2019, 54(4): 705-712 (doi: 10.15389/agrobiology.2019.4.705rus).
- Pryce J.E., Johnston J., Hayes B.J., Sahana G., Weigel K.A., McParland S., Spurlock D., Krattenmacher N., Spelman R.J., Wall E., Calus M.P.L. Imputation of genotypes from low density (50,000 markers) to high density (700,000 markers) of cows from research herds in Europe, North America, and Australasia using 2 reference populations. Journal of Dairy Science, 2014, 97(3): 1799-1811 (doi: 10.3168/jds.2013-7368).
- Ferenčaković M., Sölkner J., Curik I. Estimating autozygosity from high-throughput information: effects of SNP density and genotyping errors. Genetics Selection Evolution, 2013, 45: 42 (doi: 10.1186/1297-9686-45-42).
- Игнатьева Л.П., Белоус А.А., Недашковский И.С., Костюнина О.В., Сермягин А.А., Зиновьева Н.А. Оценка индивидуального уровня гомозиготности быков на основе геномной информации. Сибирский вестник сельскохозяйственной науки, 2019, 49(6): 79-87 (doi: 10.26898/0370-8799-2019-6-9).
- Бакоев C.Ю., Гетманцева Л.В. Методы оценки инбридинга и подписей селекции сельскохозяйственных животных на основе протяженных гомозиготных областей. Достижения науки и техники АПК, 2019, 33(11): 63-68 (doi: 10.24411/0235-2451-2019-11114).
- Смарагдов М.Г., Кудинов А.А. Полногеномная оценка инбридинга у молочного скота. Достижения науки и техники АПК, 2019, 33(6): 51-53 (doi: 10.24411/0235-2451-2019-10612).
- Сермягин А.А., Зиновьева Н.А. Генетический и геномный прогноз племенной ценности быков-производителей черно-пестрой и голштинской пород в России. Достижения науки и техники АПК, 2019, 33(12): 77-82 (doi: 10.24411/0235-2451-2019-11216).
- Chang C.C., Chow C.C., Tellier L.CAM., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 2015, 4: 7 (doi: 10.1186/s13742-015-0047-8).
- Zhang L., Orloff M.S., Reber S., Li S., Zhao Y., Eng C. aTOH: Extended Approach for Identifying Tracts of Homozygosity. PLoS ONE, 2013, 8(3): e57772 (doi: 10.1371/journal.pone.0057772).
- Hu Z.-L., Park C.A., Reecy J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Research, 2019, 47: D701-710 (doi: 10.1093/nar/gky1084).
- VanRaden P.M. Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008, 91(11): 4414-4423 (doi: 10.3168/jds.2007-0980).
- Jiang J., Ma L., Prakapenka D., VanRaden P.M., Cole J.B., Da Y. A large-scale genome-wide association study in U.S. Holstein cattle. Front. Genet., 2019, 10: 412 (doi: 10.3389/fgene.2019.00412).
- Sun J., Shan L., Zhang C., Chen H. Haplotype combination of the bovine PCSK1 gene sequence variants and association with growth traits in Jiaxian cattle. JournalofGenetics, 2014, 93(3): e123-e129. Режим доступа: http://www.ias.ac.in/jgenet/OnlineResources/93/e123.pdf. Дата обращения: 27.01.2020 года.
- Friedrich J., Brand B., Ponsuksili S., Graunke K.L., Langbein J., Knaust J., Kühn C., Schwerin M. Detection of genetic variants affecting cattle behaviour and their impact on milk production: a genome-wide association study. Anim. Genet., 2016, 47(1): 12-18 (doi: 10.1111/age.12371).
- Karisa B.K., Thomson J., Wang Z., Stothard P., Moore S.S., Plastow G.S. Candidate genes and single nucleotide polymorphisms associated with variation in residual feed intake in beef cattle. Journal of Animal Science, 2013, 91(8): 3502-3513 (doi: 10.2527/jas.2012-6170).
- Liu Y., Zan L., Xin Y., Tian W., Li L., Wang H. ZBTB38 gene polymorphism associated with body measurement traits in native Chinese cattle breeds. Gene, 2013, 513(2): 272-277 (doi: 10.1016/j.gene.2012.10.026).
- Valente T.S., Baldi F., Sant'Anna A.C., Albuquerque L.G., Paranhos da Costa M.J. Genome-wide association study between single nucleotide polymorphisms and flight speed in Nellore cattle. PLoS ONE, 2016, 11(6): e0156956 (doi: 10.1371/journal.pone.0156956).
- Lima A.O.D., Oliveira P.S.N., Tizioto P.C., Somavilla A.L., Diniz W.J.S., Silva J.V.D., Andrade S.C.S., Boschiero C., Cesar A.S.M., Souza M.M., Rocha M.I.P., Afonso J., Buss C.E., Mudadu M.A., Mourao G.B., Coutinho L.L., Regitano L.C.A. PRUNE2 gene has a potential effect on residual feed intake in Nellore cattle. Journal of Animal Science, 2016,94(E-Suppl. 5): 152-153 (doi: 10.2527/jam2016-0318).
- Cole J.B., Wiggans G.R., Ma L., Sonstegard T.S., Lawlor T.J., Crooker B.A., Van Tassell C.P., Yang J., Wang S., Matukumalli L.K., Da Y. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics, 2011, 12: 408 (doi: 10.1186/1471-2164-12-408).
- Szewczuk M. Polymorphism in exon 2 encoding the putative ligand-binding pocket of the bovine insulin-like growth factor 1 receptor affects milk traits in four different cattle breeds. Journal of Animal Breeding and Genetics, 2017, 134(1): 34-42 (doi: 10.1111/jbg.12216).
- Leal-Gutiérrez J.D., Elzo M.A., Johnson D.D., Hamblen H., Mateescu R.G. Genome wide association and gene enrichment analysis reveal membrane anchoring and structural proteins associated with meat quality in beef. BMC Genomics, 2019, 20(1): 151 (doi: 10.1186/s12864-019-5518-3).
- Hayashi K., Ushizawa K., Hosoe M., Takahashi T. Differential gene expression of serine protease inhibitors in bovine ovarian follicle: possible involvement in follicular growth and atresia. Reproductive Biology and Endocrinology, 2011, 9: 72 (doi: 10.1186/1477-7827-9-72).
- Zhuang Z., Xu L., Yang J., Gao H., Zhang L., Gao X., Li J., Zhu B. Weighted single step genome-wide association study for growth traits in Chinese Simmental beef cattle. Genes, 2020, 11: 189 (doi: 10.3390/genes11020189).
- Zhang K., Rajput S.K., Wang S., Folger J.K., Knott J.G., Smith G.W. CHD1 regulates deposition of histone variant H3.3 during bovine early embryonic development. Biology of Reproduction, 2016, 94(6): 140, 1-8 (doi: 10.1095/biolreprod.116.138693).
- Pryce J.E., Hayes B.J., Bolormaa S., Goddard M.E. Polymorphic regions affecting human height also control stature in cattle. Genetics, 2011, 187(3): 981-984 (doi: 10.1534/genetics.110.123943).
- Twomey A.J., Berry D.P., Evans R.D., Doherty M.L., Graham D.A., Purfield D.C. Genome-wide association study of endo-parasite phenotypes using imputed whole-genome sequence data in dairy and beef cattle. Genetics Selection Evolution, 2019, 51: 15 (doi: 10.1186/s12711-019-0457-7).
- Wang X.G., Ju Z.H., Hou M.H., Jiang Q., Yang C.H., Zhang Y., Sun Y., Li R.L., Wang C.F., Zhong J.F., Huang J.M. Deciphering transcriptome and complex alternative splicing transcripts in mammary gland tissues from cows naturally infected with Staphylococcus aureus mastitis. PLoS ONE, 2016, 11(12): e0167666 (doi: 10.1371/journal.pone.0167666).
- Edea Z., Jeoung J.H., Shin S.S., Ku J., Seo S., Kim I.H., Kim S.W., Kim K.S. Genome-wide association study of carcass weight in commercial Hanwoo cattle. Asian-Australasian Journal of Animal Sciences (AJAS), 2018, 31(3): 327-334 (doi: 10.5713/ajas.17.0276).
- Utsunomiya Y.T., Milanesi M., Utsunomiya A.T.H., Torrecilha R.B.P., Kim E.S., Costa M.S., Aguiar T.S., Schroeder S., do Carmo A.S., Carvalheiro R., Neves H.H.R., Padula R.C.M., Sussai T.S., Zavarez L.B., Cipriano R.S., Caminhas M.M.T., Hambrecht G., Colli L., Eufemi E., Ajmone-Marsan P., Cesana D., Sannazaro M., Buora M., Morgante M., Liu G., Bickhart D., Van Tassell C.P., Sölkner J., Sonstegard T.S., Garcia J.F. A PLAG1 mutation contributed to stature recovery in modern cattle. Scientific Reports, 2017, 7(1): 17140 (doi: 10.1038/s41598-017-17127-1).
- Li C., Sun D., Zhang S., Liu L., Alim M.A., Zhang Q. A post-GWAS confirming the SCD gene associated with milk medium- and long-chain unsaturated fatty acids in Chinese Holstein population. Animal Genetics, 2016, 47(4): 483-490 (doi: 10.1111/age.12432).
- Hering D.M., Olenski K., Kaminski S. Genome-wide association study for poor sperm motility in Holstein-Friesian bulls. Animal Reproduction Science, 2014, 146(3-4): 89-97 (doi: 10.1016/j.anireprosci.2014.01.012).
- Santos D.J.A., Cole J.B., Null D.J., Byrem T.M., Ma L. Genetic and nongenetic profiling of milk pregnancy-associated glycoproteins in Holstein cattle. Journal of Dairy Science, 2018, 101(11): 9987-10000 (doi: 10.3168/jds.2018-14682).
- Ibeagha-Awemu E.M., Akwanji K.A., Beaudoin F., Zhao X. Associations between variants of FADS genes and omega-3 and omega-6 milk fatty acids of Canadian Holstein cows. BMC Genetics, 2014, 15: 25 (doi: 10.1186/1471-2156-15-25).